Version 1.0
October 1999
![]()
![]()
(ASC) Flash Center
University of Chicago
Version 1.0
October 1999
![]()
![]()
(ASC) Flash Center
University of Chicago
The FLASH Code Group is supported by the (ASC) Flash Center at the University of Chicago under U. S. Department of Energy contract B341495. Some of the test calculations described here were performed using the Origin 2000 computer at Argonne National Laboratory and the (ASC) Blue Mountain computer at Los Alamos National Laboratory.
The Center for Astrophysical Thermonuclear Flashes, or Flash Center, was founded at the University of Chicago in 1997 under contract to the United States Department of Energy as part of its Accelerated Strategic Computing Initiative ((ASC)). The goal of the Center is to solve several problems related to thermonuclear flashes on the surfaces of compact stars (neutron stars and white dwarfs), in particular X-ray bursts, Type Ia supernovae, and novae. To solve these problems requires the participants in the Center to develop new simulation tools capable of handling the extreme resolution and physical requirements imposed by conditions in these explosions, and to do so while making efficient use of the parallel supercomputers developed by the ASC project, the most powerful constructed to date.
The FLASH code represents an important step along the road to this goal. FLASH is a modular, adaptive, parallel simulation code capable of handling general compressible flow problems in astrophysical environments. FLASH has been designed to allow users to configure initial and boundary conditions, change algorithms, and add new physical effects with minimal effort. It uses the PARAMESH library to manage a block-structured adaptive grid, placing resolution elements only where they are needed most. FLASH uses the Message-Passing Interface (MPI) library to achieve portability and scalability on a variety of different message-passing parallel computers.
This user's guide is designed to enable individuals unfamiliar with the FLASH code to quickly get acquainted with its structure and to move beyond the simple test problems distributed with FLASH, customizing it to suit their own needs. The second section briefly describes the equations and algorithms used by the physics modules distributed with FLASH. It assumes that the reader has some familiarity both with the basic physics involved and with numerical hydrodynamics methods. This familiarity is absolutely essential in using FLASH (or any other simulation code) to arrive at meaningful solutions to physical problems. The novice reader is directed to an introductory text such as Patrick Roache's Fundamentals of Computational Fluid Dynamics (Hermosa, 1998) or C. A. J. Fletcher's Computational Techniques for Fluid Dynamics (Springer-Verlag, 1991). The advanced reader who wishes to know more specific information about the various algorithms is directed to the literature references in this section.
The third section discusses how to quickly get started with FLASH, describing how to configure, build, and run the code with one of the included test problems, then examine the resulting output. Users familiar with the capabilities of FLASH who wish to quickly `get their feet wet' with the code can skip directly to this section. The fourth section describes in more detail the use of the configuration and analysis tools distributed with FLASH. The fifth section describes the different test problems distributed with FLASH. The sixth section describes the FLASH code structure at length and gives detailed instructions for extending FLASH's capabilities by adding new problem setups. This section also discusses the code modules included with FLASH 1.0 and describes how new solvers may be integrated into the code. Finally, the seventh section gives guidance on contacting the authors of FLASH.
FLASH provides a general simulation framework capable of incorporating several different types of physics module (e.g., hydrodynamics on structured or unstructured meshes, N-body solvers, reactive source terms, etc.) and initial or boundary conditions. However, it has been built to solve a particular class of problems, namely reactive, compressible flows in astrophysical environments. Thus, FLASH 1.0 is distributed with a particular set of algorithms to handle compressible hydrodynamics on a block-structured adaptive mesh, together with an appropriate nuclear equation of state and nuclear reaction network. We treat each of these elements in turn.
The hydrodynamic module in FLASH 1.0 is based on the PROMETHEUS code (Fryxell, Müller, and Arnett 1989). This code solves Euler's equations for compressible gas dynamics in one, two, or three spatial dimensions. These equations can be written in conservative form as
| |||||||||||||||||||||||||||
| (4) |
| (5) |
For reactive flows, a separate advection equation must be solved for each chemical or nuclear species:
| (6) |
The equations are solved using a modified version of the Piecewise-Parabolic Method (PPM), which is described in detail in Woodward and Colella (1984) and Colella and Woodward (1984). PPM is a higher-order version of the method developed by Godunov (1959). Godunov's method uses a finite-volume spatial discretization of the Euler equations together with an explicit forward time difference. Time-advanced fluxes at cell boundaries are computed using the analytic solution to Riemann's shock tube problem at each boundary. Initial conditions for each Riemann problem are determined by assuming the nonadvanced solution to be piecewise constant in each cell. Using the Riemann solution has the effect of introducing explicit nonlinearity into the difference equations and permits the calculation of sharp shock fronts and contact discontinuities without introducing significant nonphysical oscillations into the flow. Since the value of each variable in each cell is assumed to be constant, Godunov's method is limited to first-order accuracy in both space and time.
PPM improves on Godunov's method by representing the flow variables with piecewise parabolic functions. It also uses a monotonicity constraint rather than artificial viscosity to control oscillations near discontinuities, a feature shared with the MUSCL scheme of van Leer (1979). Although this could lead to a method which is accurate to third order, PPM is formally accurate only to second order in both space and time, as a fully third-order scheme proved not to be cost-effective. Nevertheless, PPM is considerably more accurate and efficient than most formally second-order algorithms.
PPM is particularly well-suited to flows involving discontinuities, such as shocks and contact discontinuities. The method also performs extremely well for smooth flows, although other schemes which do not perform the extra work necessary for the treatment of discontinuities might be more efficient in these cases. The high resolution and accuracy of PPM are obtained by the explicit nonlinearity of the scheme and through the use of intelligent dissipation algorithms, such as monotonicity enforcement, contact steepening, and interpolant flattening. These algorithms are described in detail by Colella and Woodward (1984).
A complete description of PPM is beyond the scope of this user's guide. However, for comparison with other codes, we note that the implementation of PPM in FLASH 1.0 uses the direct Eulerian formulation of PPM and the technique for allowing nonideal equations of state described by Colella and Glaz (1985). For multidimensional problems, FLASH 1.0 uses second-order operator splitting (Strang 1968). FLASH also implements PPM on a block-structured adaptive mesh, which is described in the following section.
We have used a package known as PARAMESH (MacNeice et al. 1999) for the parallelization and adaptive mesh refinement (AMR) portion of FLASH. PARAMESH consists of a suite of subroutines which handle refinement/derefinement, distribution of work to processors, guard cell filling, and flux conservation. In this section we briefly describe this package and the ways in which it has been modified for use with FLASH.
PARAMESH uses a block-structured adaptive mesh refinement scheme similar to others in the literature (e.g., Parashar 1999; Berger & Oliger 1984; Berger & Colella 1989; DeZeeuw & Powell 1993) as well as to schemes which refine on an individual cell basis (Khokhlov 1997). In block-structured AMR, the fundamental data structure is a block of uniform cells arranged in a logically Cartesian fashion. Each cell can be specified using a block identifier (processor number and local block number) and a coordinate triple (i,j,k), where i = 1�nxb, j = 1�nyb, and k = 1�nzb. The complete computational grid consists of a collection of blocks with different physical cell sizes, related to each other in a hierarchical fashion using a tree data structure. The blocks at the root of the tree have the largest cells, while their children have smaller cells and are said to be refined. Two rules govern the establishment of refined child blocks in PARAMESH. First, the cells of a refined child block must be one-half as large as those of its parent block. Second, a block's children must be nested; that is, the child blocks must fit within their parent block and cannot overlap one another, and the complete set of children of a block must fill its volume. Thus, in d dimensions a given block can have at most 2d children.
Each block contains nxb×nyb×nzb interior cells and a set of guard cells. The guard cells contain boundary information needed to update the interior cells. These can be obtained from physically neighboring blocks, externally specified boundary conditions, or both. The number of guard cells needed depends upon the interpolation scheme and differencing stencil used for the hydrodynamics algorithm; for the explicit PPM algorithm distributed with FLASH, four guard cells are needed in each direction. PARAMESH handles the filling of guard cells with information from other blocks or a user-specified external boundary routine. If a block's neighbor has the same level of refinement, PARAMESH fills its guard cells using a direct copy from the neighbor's interior cells. If the neighbor has a different level of refinement, the neighbor's interior cells are used to interpolate guard cell values for the block. If the block and its neighbor are stored in the memory of different processors, PARAMESH handles the appropriate parallel communication (blocks are not split between processors). PARAMESH supports only linear interpolation for guard cell filling at jumps at refinement, but it is easily extended to allow other interpolation schemes; for example, for FLASH we have added a quadratic interpolation scheme. Once each block's guard cells are filled, it can be updated independently of the other blocks.
PARAMESH also enforces flux conservation at jumps in refinement, as described by Berger and Colella (1989). At jumps in refinement, the fluxes of mass, momentum, energy, and nuclear abundances across boundary cell faces are averaged across the fine cells at that face and provided to the corresponding coarse face on the neighboring block. These averaged fluxes are then used to update the values of the coarse block's local flow variables.
Each processor decides when to refine or derefine its blocks by computing a user-defined error estimator for each block. Refinement involves creation of between one and 2d refined child blocks, while derefinement involves deletion of a block. As child blocks are created, they are temporarily placed at the end of the processor's block list. After the refinements and derefinements are complete, the blocks are redistributed among the processors using a work-weighted Morton space-filling curve in a manner similar to that described by Warren and Salmon (1987) for a parallel treecode. During the distribution step each block is assigned a work value (an estimate of the relative amount of time required to update the block). The Morton number of the block is then computed by interleaving the bits of its integer coordinates as described by Warren and Salmon (1987); this determines its location along the space-filling curve. The Morton numbers of all of the blocks in the problem are then sorted using a parallel bitonic sort. Finally, the list of all blocks is partitioned among the processors using the block weights, equalizing the estimated workload of each processor. During the sorting step, only Morton numbers and not block data are communicated between processors; blocks are only moved (if necessary) after the sort. Changes in refinement are typically performed every ten timesteps.
The refinement criterion used by PARAMESH is adapted from Löhner (1987). Löhner's error estimator was originally developed for finite element applications and has the advantage that it uses an entirely local calculation. Furthermore, the estimator is dimensionless and can be applied with complete generality to any of the field variables of the simulation or any combination of them (by default, PARAMESH uses the density and pressure). Löhner's estimator is a modified second derivative, normalized by the average of the gradient over one computational cell. In one dimension on a uniform mesh it is given by
| (7) |
| (8) |
It is common to call an equation of state routine more than 109 times when calculating two- and three-dimensional hydrodynamic models of stellar phenomena. Thus, it is very desirable to have an equation of state that is as efficient as possible, yet accurately represents the relevant physics. While FLASH is easily capable of including any equation of state, here we discuss three equation of state (henceforth EOS) routines which are supplied with FLASH 1.0: the ideal-gas or gamma-law EOS, the Nadyozhin EOS, and the Helmholtz EOS.
Each EOS routine takes as input the temperature T, density r, mean number of nucleons per isotope [`A], and mean charge per isotope [`Z]. Each routine then returns as output the scalar pressure (Ptot, specific thermal energy etot, and entropy Stot. In addition to these quantities, the EOS routines return partial derivatives of the pressure, specific thermal energy, and entropy with respect to the density and temperature:
| (9) |
An EOS is thermodynamically consistent if its outputs satisfy the Maxwell relations:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||
The gamma-law EOS provided with FLASH represents a simple ideal gas with constant adiabatic index g:
| (13) |
More complex equations of state are necessary for realistic models of X-ray bursts, Type Ia supernovae, classical novae, and other stellar phenomena, where the electrons and positrons may be relativistic and/or degenerate and where radiation contributes significantly to the thermodynamic state. The Nadyozhin and Helmholtz EOS routines address this need. The Nadyozhin EOS, summarized by Nadyozhin (1974) and explained in detail by Blinnikov et al. (1996), was found by Timmes and Arnett (1999) in a comparison of five different analytic EOS schemes to give the best tradeoff among accuracy, thermodynamic consistency, and speed. The Helmholtz EOS (Timmes & Swesty 1999) uses interpolation from a two-dimensional table of the Helmholtz free energy F(r,T), obtaining comparable accuracy, better thermodynamic consistency, and about twice the speed of the Nadyozhin EOS.
For stellar phenomena, the pressure, specific thermal energy, and entropy contain contributions from several components:
| |||||||||||||||||||||
| (17) |
| |||||||||||||||||||
| (20) |
| (21) |
| (22) |
The Nadyozhin EOS routine uses polynomial or rational functions to evaluate the thermodynamic quantities. The temperature-density plane is decomposed into 5 regions (see Figure 12 of Blinnikov et al. 1996), with different expansions or fitting functions applied to each region. The methods used in the 5 regions are (1) a perfect gas approximation with the first-order corrections for degeneracy, (2) expansions of the half-integer Fermi-Dirac functions, (3) Chandrasekhar's (1939) expansion for a degenerate gas, (4) relativistic asymptotics, and (5) Gaussian quadrature for the thermodynamic quantities. The perturbation expansions, asymptotic relations, and fitting functions for the 5 regions are combined, with extraordinary care given to making sure that transitions between the regions are continuous, smooth, and thermodynamically consistent. All of the partial derivatives are obtained analytically.
The electron-positron Helmholtz free energy table used by the Helmholtz EOS was generated by using the Timmes EOS routine (Timmes & Arnett 1999). The Fermi-Dirac integrals, along with their derivatives with respect to h and b, were calculated to at least 18 significant figures using the quadrature schemes of Aparicio (1998). Newton-Raphson iteration was used to solve for h to at least 15 significant figures. The thermodynamic derivatives were computed analytically. Searches through the free energy table are avoided by computing hash indices from the values of any given (T,r[`Z]/[`A]) pair. No computationally expensive divisions are required in interpolating from the table; all of them can be computed and stored the first time the EOS routine is called.
Modelling thermonuclear flashes typically requires the energy generation rate due to nuclear burning over a large range of temperatures, densities and compositions. The average energy generated or lost over a period of time is found by integrating a system of ordinary differential equations (the nuclear reaction network) for the abundances of important nuclei and the total energy release. In some contexts, such as Type II supernova models, the abundances themselves are also of interest. In either case, the coefficients that appear in the equations typically are extremely sensitive to temperature. The resulting stiffness of the system of equations requires the use of an implicit time integration scheme.
Timmes (1999) has reviewed several methods for solving stiff nuclear reaction networks, providing the basis for the reaction network solver included with FLASH. The scaling properties and behavior of three semi-implicit time integration algorithms (a traditional first-order accurate Euler method, a fourth-order accurate Kaps-Rentrop method, and a variable order Bader-Deuflhard method) and eight linear algebra packages (LAPACK, LUDCMP, LEQS, GIFT, MA28, UMFPACK, and Y12M) were investigated by running each of these 24 combinations on seven different nuclear reaction networks (hard-wired 13- and 19-isotope networks and table-lookup networks using 47, 76, 127, 200, and 489 isotopes). Timmes' analysis suggested that the best balance of accuracy, overall efficiency, memory footprint, and ease-of-use was provided by the choice of a 13- or 19-isotope reaction network, the variable order Bader-Deuflhard time integration method, and the MA28 sparse matrix package. The default FLASH distribution uses this combination of methods, which we describe in this section.
We begin by describing the equations solved by the nuclear burning module. We consider material which may be described by a density r and a single temperature T and contains a number of isotopes i, each of which has Zi protons and Ai nucleons (protons + neutrons). Let ni and ri denote the number and mass density, respectively, of the ith isotope, and let Xi denote its mass fraction, so that
| (23) |
| (24) |
| (25) |
The general continuity equation for the ith isotope is given in Lagrangian formulation by
| (26) |
| (27) |
The case Vi � 0 is important for two reasons. First, mass diffusion is often unimportant when compared to other transport process such as thermal or viscous diffusion (i.e., large Lewis numbers and/or small Prandtl numbers). Such a situation obtains, for example, in the study of laminar flame fronts propagating through the quiescent interior of a white dwarf. Second, this case permits the decoupling of the reaction network solver from the hydrodynamical solver through the use of operator splitting, greatly simplifying the algorithm. This is the method used by the default FLASH distribution. Setting Vi � 0 transforms equation (26) into
| (28) |
| (29) |
Because of the highly nonlinear temperature dependence of the nuclear reaction rates, and because the abundances themselves often range over several orders of magnitude in value, the values of the coefficients which appear in equations (28) and (29) can vary quite significantly. As a result, the nuclear reaction network equations are ``stiff.'' A system of equations is stiff when the ratio of the maximum to the minimum eigenvalue of the Jacobian matrix [(J)\tilde] � �f/�y is large and imaginary. This means that at least one of the isotopic abundances changes on a much shorter timescale than another. Implicit or semi-implicit time integration methods are generally necessary to avoid following this short-timescale behavior, requiring the calculation of the Jacobian matrix.
It is instructive at this point to look at an example of how equation (28) and the associated Jacobian matrix are formed. Consider the 12C(a,g)16O reaction, which competes with the triple-a reaction during helium burning in stars. The rate R at which this reaction proceeds is critical for evolutionary models of massive stars since it determines how much of the core is carbon and how much of the core is oxygen after the initial helium fuel is exhausted. This reaction sequence contributes to the right-hand side equation (29) through the terms
| |||||||||||||||||||||
| |||||||||||||||
The default FLASH distribution includes two reaction networks. The 13-isotope a-chain plus heavy-ion reaction network is suitable for most multi-dimensional simulations of stellar phenomena where having a reasonably accurate energy generation rate is of primary concern. The 19-isotope reaction network has the same a-chain and heavy-ion reactions as the 13-isotope network, but it includes additional isotopes to accommodate some types of hydrogen burning (PP and CNO chains), along with some aspects of photodisintegration into 54Fe. This reaction network is described in Weaver, Zimmerman, & Woosley (1978). Both the networks supplied with FLASH are examples of ``hard-wired'' reaction networks, where each of the reaction sequences are carefully entered by hand. This approach is suitable for small networks when minimizing the CPU time required to run the reaction network is a primary concern, although it suffers the disadvantage of inflexibility.
The Jacobian matrices of nuclear reaction networks tend to be sparse, and they become more sparse as the number of isotopes increases. Implicit time integration schemes require the inverse of the Jacobian matrix, so we need to use a sparse linear algebra package to compute this inverse. To control the iteration count for a prespecified accuracy, FLASH uses a direct sparse matrix solver. Direct methods typically divide the solution of [(A)\tilde] ·x = b into a symbolic LU decomposition, numerical LU decomposition, and a backsubstitution phase. In the symbolic LU decomposition phase the pivot order of a matrix is determined, and a sequence of decomposition operations which minimize the amount of fill-in is recorded. Fill-in refers to zero matrix elements which become nonzero (e.g., a sparse matrix times a sparse matrix is generally a denser matrix). The matrix is not (usually) decomposed; only the steps to do so are stored. Since the nonzero pattern of a chosen nuclear reaction network does not change, the symbolic LU decomposition is a one-time initialization cost for reaction networks. In the numerical LU decomposition phase, a matrix with the same pivot order and nonzero pattern as a previously factorized matrix is numerically decomposed into its lower-upper form. This phase must be done only once for each staged set of linear equations. In the backsubstitution phase, a set of linear equations is solved with the factors calculated from a previous numerical decomposition. The backsubstitution phase may be performed with as many right-hand sides as needed, and not all of the right-hand sides need to be known in advance.
The MA28 linear algebra package used by FLASH is described by Duff, Erisman, & Reid (1986). It uses a combination of nested dissection and frontal envelope decomposition to minimize fill-in during the factorization stage. An approximate degree update algorithm that is much faster (asymptotically and in practice) than computing the exact degrees is employed. One continuous real parameter sets the amount of searching done to locate the pivot element. When this parameter is set to zero, no searching is done and the diagonal element is the pivot, while when set to unity, complete partial pivoting is done. Since the matrices generated by reaction networks are usually diagonally dominant, the routine is set in FLASH to use the diagonal as the pivot element. Several test cases showed that using partial pivoting did not make a significant accuracy difference, but were less efficient since a search for an appropriate pivot element had to be performed. MA28 accepts the nonzero entries of the matrix in the (i, j, ai,j) coordinate system, and typically uses uses 70-90% less storage than storing the full dense matrix.
The time integration method used by FLASH for evolving the reaction networks is the variable order Bader-Deuflhard method (e.g., Bader & Deuflhard 1983; Press et al. 1992). The reaction network is advanced over a large time step H from yn to yn+1 by the following sequence of matrix equations. First,
| |||||||||||||||||
| |||||||||||||||||
| ||||||||||||||
The energy generation rate is given by the sum
| (35) |
| (36) |
Finally, the tabulation of Caughlan & Fowler (1988) is used in FLASH for most of the key nuclear reaction rates. Modern values for some of the reaction rates were taken from the reaction rate library of Hoffman (1999, priv. comm.). Nuclear reaction rate screening effects as implemented by Wallace, Woosley, & Weaver (1982), and decreases in the energy generation rate [(e)\dot]nuc due to neutrino losses as given by Itoh et al. (1985) are included in calculations done with FLASH.
This section describes how to quickly get up and running with FLASH, showing how to configure and build it to solve the Sedov explosion problem, how to run it, and how to examine the output using IDL. To begin, verify that you have the following:
FLASH has been tested on the following Unix-based platforms. In addition, it may work with others not listed (see section ).
In this section we will use irix as an example of the machine type. Substitute your machine type for irix wherever it appears.
To begin, unpack the FLASH source code distribution. If you have a Unix tar file, type `tar xvf FLASHX.tar' (without the quotes), where X is the FLASH major version number (for example, use FLASH1.tar and FLASH1/ for FLASH version 1.0). If you are working with a CVS source tree, use `cvs checkout FLASHX' to obtain a personal copy of the tree. You may need to obtain permission from the local CVS administrator to do this. In either case you will create a directory called FLASHX/. Type `cd FLASHX' to enter this directory.
Next, configure the FLASH source tree for the Sedov explosion problem. Type
./setup sedov irix -defaults
This configures FLASH for the sedov problem, using the default hydrodynamic solver, equation of state, and I/O format defined for this problem. The source tree is configured to create a two-dimensional code by default.
The next step is to edit the file object/Makefile.h. This file contains all of the site-dependent and architecture-dependent parts of the Makefile. Verify that the compiler settings (FCOMP, CCOMP, and CPPCOMP) and the library paths (LIB) are correct for your system. No other changes should be necessary at this stage (later on you may wish to fiddle with the compiler optimization settings). Future versions of FLASH may make use of the GNU configure program to handle this step.
From the FLASH root directory (ie. the directory from which you ran setup), execute make. This will compile the FLASH code. If you should have problems and need to recompile, `make clean' will remove all object files from the object/ directory, leaving the source configuration intact; `make realclean' will remove all files and links from object/. After `make realclean,' a new invocation of setup is required before the code can be built.
Assuming compilation and linking were successful, you should now find an executable named flashX_sedov_irix in the object/ directory. (Remember to replace irix with your machine type.) You may wish to check that this is the case.
FLASH expects to find a flat text file named flash.par in the directory from which it is run. This file sets the values of various runtime parameters which determine the behavior of FLASH. If it is not present, FLASH will use default values for the runtime parameters, which may or may not produce desirable results. Here we will create a simple flash.par which sets a few parameters and allows the rest to take on default values. With your text editor, create flash.par in the main FLASH directory with the contents of Figure 1.
# runtime parameterslrefine_max = 4
basenm = ßedov_4_"
restart = .false.
trstrt = 0.005
nend = 1000
tmax = 0.02
gamma = 1.4
xlboundary = -21 # code -21 is outflow
xrboundary = -21
ylboundary = -21
yrboundary = -21
This instructs FLASH to use up to four levels of adaptive mesh refinement (AMR) and to name the output files appropriately. We will not be starting from a checkpoint file (this is the default, but here it is explicitly set for purposes of illustration). Output files are to be written every 0.005 time units, and we will integrate until t = 0.02 or 1000 timesteps have been taken, whichever comes first. The ratio of specific heats for the gas (g) is taken to be 1.4, and all four boundaries of the two-dimensional grid have outflow (zero-gradient or Neumann) boundary conditions. Note the format of the file: each line is a comment (denoted by a hash mark, #), blank, or of the form variable = value. String values are enclosed in double quotes ("). Boolean values are indicated in the Fortran style, .true. or .false.
We are now ready to run FLASH. To run FLASH on N processors, type
mpirun -np N object/flashX_sedov_irix
remembering to replace N, X, and irix with the appropriate values. Some systems may require you to start MPI programs with a different command; use whichever command is appropriate to your system. The FLASH executable can take one command-line argument, the name of the runtime parameter file. The default parameter file name is flash.par.
You should see a number of lines of output indicating that FLASH is initializing the Sedov problem, listing the initial parameters, and giving the timestep chosen at each step. At the end FLASH prints a summary of the CPU time spent in various parts of the code and terminates. In the current directory you should now find several files:
We will use the xflash routine under IDL to examine the output. Before doing so, we need to set the values of two environment variables, IDL_PATH and XFLASH_DIR. Under csh this can be done using the commands
setenv XFLASH_DIR "$PWD/tools/idl"
setenv IDL_PATH "${XFLASH_DIR}:$IDL_PATH"
If you get a message indicating that IDL_PATH is not defined, just enter
setenv IDL_PATH "$XFLASH_DIR"
Now run IDL (idl) and enter xflash at the IDL> prompt. You should see a control panel widget as shown in Figure 2.
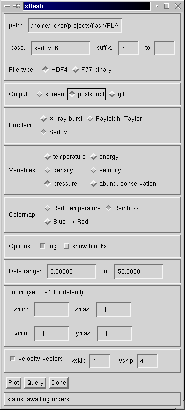
The path entry should be filled in for you with the current directory. Enter sedov_4_ as the base filename and enter 4 as the suffix. (xflash can generate output for a number of consecutive files, but if you fill in only the beginning suffix, only one file is read.) Our output files are in HDF format, so make sure the HDF button is selected. Choose the image format (screen, Postscript, GIF) and the problem type (in our case, Sedov). Selecting the problem type is only important for choosing default ranges for our plot; plots for other problems can be generated by ignoring this setting and overriding the default values for the data range and the coordinate ranges. Select the desired plotting variable and colormap. The `abundance conservation' variable produces a plot of the sum of the nuclear abundances, useful for comparison with the expected value (unity). Under `Options,' select whether to plot the logarithm of the desired quantity, and select whether to plot the outlines of the AMR blocks. For very highly refined grids the block outlines can obscure the data, but they are useful for verifying that FLASH is putting resolution elements where they are needed. Finally, check `velocity vectors' to overlay the velocity field. The `xskip' and `yskip' parameters enable you plot only a fraction of the vectors so that they do not obscure the background plot.
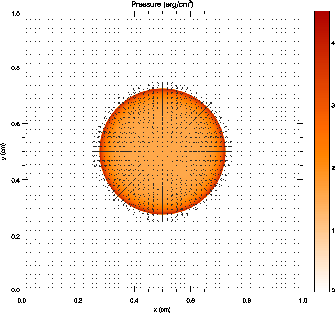
When the control panel settings are to your satisfaction, click the `Plot' button to generate the plot. For Postscript and GIF output, a file is created in the current directory. The result should look something like Figure 3, although this figure was generated from a run with eight levels of refinement rather than the four used in the quick start example run. With fewer levels of refinement the Cartesian grid causes the explosion to appear somewhat diamond-shaped.
FLASH is intended to be customized by the user to work with interesting initial and boundary conditions. In the following sections we will cover in more detail the structure of FLASH and the sample problems and tools distributed with it.
In this section we describe several of the tools distributed with FLASH. The source code for these programs (except for setup) can be found in the main FLASH source tree under the tools/ directory.
The setup script, found in the FLASH root directory, provides the primary command-line interface to the FLASH source code. It configures the source tree for a given problem and target machine and creates files needed to parse the runtime parameter file and make the FLASH executable. More description of what setup does may be found in Section 6.1. Here we describe its basic usage.
Running setup without any options prints a message describing the available options:
Available values for the two mandatory options, the name of the problem to configure and the type of machine to configure for, are determined by scanning the setups/ and source/systems/ directories, respectively.[sphere 5:09pm] % ./setup
usage: setup problem-name machine-type [options]
problems: 2blast advect mach rt sedov sod xrayburst
machines: asci_blue asci_bluemtn asci_red irix linux t3e
options: -verbose -portable -defaults -[123]d -maxblocks=#
A ``problem'' consists of a set of initial and boundary conditions, possibly additional physics (e.g., a subgrid model for star formation), and a set of adjustable parameters. The directory associated with a problem contains source code files which implement the initial and boundary conditions and extra physics, together with a configuration file, read by setup, which contains information on required physics modules and adjustable parameters.
A ``machine type'' consists of an architecture and an associated operating system. Normally it does not refer to a specific machine, but in the case of the (ASC) machines (Blue Pacific, Blue Mountain, and Red), the particular machine is unique and is treated as a separate architecture. The directory for each machine type contains a makefile fragment ( Makefile.h) which sets command names, compiler flags, and library paths, and any replacement or additional source files needed to compile FLASH for that machine type.
setup uses the problem and machine type, together with a user-supplied file called Modules which lists the code modules to include, to generate a directory called object/ which contains links to the appropriate source files and makefile fragments. It also creates the master makefile (object/Makefile) and several Fortran include files (object/rt_*) which are needed by the code in order to parse the runtime parameter file. After running setup, the user can make the FLASH executable by running make in the object/ directory (or from the FLASH root directory, if the -portable option is not used with setup). The optional command-line modifiers have the following interpretations:
| -verbose | Normally setup echoes to the standard output summary messages indicating what it is doing. Including the -verbose option causes it to also list the links it creates. |
| -portable | This option creates a portable build directory. Instead of object/, setup creates object_problem_machine/, and instead of creating links to the source files in source/ and setups/, it copies files from those directories into the build directory. The resulting build directory can be placed into a tar archive and sent to another machine for building. Because the makefile in the FLASH root directory is generic and does not know what problem or machine has been selected, it cannot be used to make code which has been configured with -portable. |
| -defaults | Normally setup requires that the user supply a plain text file called Modules (in the FLASH root directory) which specifies which code modules to include. A sample Modules file appears in Figure 4. Each line is either a comment (preceded by a hash mark (#)) or a module include statement of the form INCLUDE module. Sub-modules are indicated by specifying the path to the sub-module in question; in the example, the sub-module gamma of the eos module is included. If a module has a default sub-module, but no sub-module is specified, setup automatically selects the default using information provided by the module's configuration file. |
| The purpose of the -defaults option is to enable setup to generate a ``rough draft'' of a Modules file for the user. The configuration file for each problem setup specifies a number of code module requirements; for example, a problem may require the perfect-gas equation of state (eos/gamma) and an unspecified hydro solver (hydro). With -defaults, setup creates a Modules file by converting these requirements into module include statements. In addition, it checks the configuration files for the required modules and includes any of their required modules, eliminating duplicates. This is only done for one level of requirement, but usually this is enough. Most users configuring a problem for the first time will want to run setup with -defaults to generate a Modules file, then edit Modules directly to specify different sub-modules (e.g., hydro solvers or I/O formats). After editing Modules in this way, re-run setup without -defaults to incorporate the changes into the code configuration. |
# Modules file constructed for rt problem by setup -defaults
INCLUDE driver
INCLUDE eos/gamma
INCLUDE grav
INCLUDE paramesh
INCLUDE mmpi
INCLUDE mpi_amr
INCLUDE hydro
INCLUDE io
| -[123]d | By default setup creates a makefile which produces a FLASH executable capable of solving one- or two-dimensional problems (equivalent to -2d). To generate a makefile with options appropriate to three-dimensional problems, use -3d. The resulting code will be able to solve 1D, 2D, or 3D problems. To generate a one-dimensional code, use -1d. While -3d provides maximum flexibility, it also allocates the most memory at runtime. It is generally very wasteful of memory to solve a 1D or 2D problem using an executable compiled with -3d. These options are mutually exclusive and cause setup to add the appropriate compilation option to the makefile it generates. |
| -maxblocks=# | This option is also used by setup in constructing the makefile compiler options. It determines the amount of memory allocated at runtime to the adaptive mesh refinement (AMR) block data structure. For example, to allocate enough memory on each processor for 500 blocks, use -maxblocks=500. If the default block buffer size is too large for your system, you may wish to try a smaller number here (the defaults are currently defined in source/driver/physicaldata.fh). Alternatively, you may wish to experiment with larger buffer sizes if your system has enough memory. |
The FLASH output comparison utility, or focu (pronounced faux-coup), is a command-line utility which compares FLASH output files according to specified criteria (e.g., local or global error criterion on any of the hydrodynamical variables) and reports whether they are the same or not. This is useful in determining whether a change to FLASH (or to the value of a runtime parameter) has changed the results obtained with a given problem.
The focu executable is configured and built in much the same way as is FLASH: obtain source tree, run setup to configure the source tree for a given architecture, and run make in the build directory. FLASH and focu are different in that focu is problem-independent, so only a machine type needs to be specified, and in that (at least at the present time) all of the modules included with focu are used, so setup is always run with the -defaults option. (See Section 4.1 for details regarding the usage of setup.) To summarize the steps in building focu:
Additional setup options are available and are interpreted as described in Section 4.1.
Running focu is very similar to running FLASH itself. First prepare a runtime parameter file, then run focu using
(or use the command used by your system to start MPI programs). Here N is the number of processors to use (which need not be the same as the number of processors used by the FLASH job which produced the output files to be read), and machine-type is the machine type specified in the setup step. If the parameter-file argument is not supplied, focu looks for a parameter file named focu.par in the current directory. The format of this file is identical to that used by FLASH (see Section 3).mpirun -np N focu_machine-type parameter-file
The runtime parameters understood by focu are as follows. Default values are listed in brackets following each description.
| file1 | Base file name for the first file to compare. The file name will be constructed by appending the file number, padded with zeros to four places, to the base file name. [``file1''] |
| num1 | The file number for the first file to compare. [0] |
| format1 | The format of the first file: ``HDF'' for Hierarchical Data Format, ``F77'' for Fortran unformatted. [``HDF''] |
| file2 | Base file name for the second file to compare. [``file2''] |
| num2 | File number for the second file. [0] |
| format2 | Format for the second file. [``HDF''] |
| variable | The name of the hydrodynamical variable to examine. Available values are ``density,'' ``pressure,'' ``x velocity,'' ``y velocity,'' ``z velocity,'' ``temperature,'' ``energy,'' ``kinetic energy,'' ``internal energy,'' ``x momentum,'' ``y momentum,'' ``z momentum,'' and ``abundance N,'' where N is the index of the abundance to use. [``density''] |
| criterion | The error criterion to apply in comparing the files. This must be of the form ``A B'', where A is either ``local'' or ``global,'' and B is either ``sum,'' ``min,'' or ``max.'' For the global criteria, focu first computes the volume-weighted average, minimum value, or maximum value of variable for each file, then computes the L1 deviation between the two global quantities. For the local criteria, the files are compared on a zone-by-zone basis. Consequently the AMR block structures of the two files must be the same in order for the comparison to succeed. If criterion is ``local sum,'' the volume-weighted average of the local L1 deviations is compared to threshold to determine success or failure. If criterion is ``local min'' or ``local max,'' the minimum or maximum L1 deviation is compared. The L1 deviation for two quantities X1 and X2 is defined as |X2-X1| / max{1/2|X1+X2|,10-99}. [``global sum''] |
| threshold | The error threshold to use in deciding success or failure. [10-6] |
| outfile | The name of the output file to create when generating the comparison report. Setting this to ``stdout'' causes focu to write to standard output. [``stdout''] |
FOCU: Flash Output Comparison Utility
Comparing: ßedov_4_f77_" # 3 ßedov_4_hdf_" # 3 Variable: density Criterion: local sum Threshold: 1.0000000000000E-09 Result: Error: 0.0000000000000E+00 Minimum: 0.0000000000000E+00 Maximum: 0.0000000000000E+00 SUCCESS
The output of focu is a short report listing the volume-averaged sum, minimum, and maximum deviations (local or global, depending on the setting of criterion) and reporting SUCCESS or FAILURE according to the supplied threshold. For example, if two runs of FLASH are performed using the Sedov problem, one with HDF output and one with Fortran unformatted output, the resulting output files should be identical. The report generated by a focu comparison of two such files is shown in Figure 5. Note that the last line of the report begins with SUCCESS or FAILURE, making it easy for a script to test the results. For example, with csh one might use
focu is used by the automated FLASH test script (Section 5.1) to compare FLASH outputs to the reference results supplied as part of the test suite.set result = `mpirun -np 2 focu_irix focu_sedov.par | grep SUCCESS`
if ("result" == "SUCCESS") then
...
endif
fidlr is a set of routines, written in the IDL language, which provide tools for plotting results obtained with FLASH. The routines include programs which can be run from the IDL command line to read 2D or 3D FLASH datasets and interpolate them onto uniform grids. An IDL program called xflash provides a graphical interface to these routines, enabling users to read and plot 2D AMR datasets without interpolating onto a uniform mesh.
The FLASH IDL routines are located in the tools/fidlr/ subdirectory of the FLASH root directory. To use them you must set two environment variables. First set the value of XFLASH_DIR to the location of the FLASH IDL routines; for example, under csh, use
setenv XFLASH_DIR flash-root-path/tools/fidlr
where flash-root-path is the absolute path of the FLASH root directory. Next add this directory to your IDL_PATH variable:
setenv IDL_PATH "${XFLASH_DIR}:$IDL_PATH"
If you have not previously defined IDL_PATH, omit :$IDL_PATH from the above command. You may wish to include these commands in your .cshrc file (or profile if you use another shell) to avoid having to reissue them every time you log in.
To begin using the routines, start IDL (idl). You may wish to make use of the program start.pro included with fidlr; this forces the use of an eight-bit colormap on 24-bit displays which have the ability to maintain two color depths (such as those found on SGIs). To use this program, start IDL using idl start.pro.
xflash provides a graphical interface to IDL for plotting one or several 2D AMR datasets. It plots variables using a shaded colormap and can overlay the outline of the AMR block structure or arrows showing the velocity field. It also enables users to zoom in to different regions of the dataset and to probe the dataset.
To use the graphical tool, from the IDL command line (idl>)
execute xflash. The widget is divided into sections controlling
the file location, output type, problem, variable to plot, colormap, plot
options, data range, zooming, and velocity vectors. At the bottom is
a status display. See Figure 2 for an example of the
appearance of the xflash window.
File location
The files to be read are specified by entering a path (by default the
current IDL working directory), a base name, and a range of suffixes.
If only the first suffix is entered, only that one is used; otherwise
all integers between the beginning and ending suffix are used.
Select the file type by choosing ``HDF'' for Hierarchical Data Format
or ``F77'' for Fortran unformatted output. At present the Fortran
reader does not convert little-endian files to big-endian or vice versa.
Output device
It is possible to direct output to the screen, to a Postscript file, or
to a GIF image. The plotting routine
will determine the orientation (portrait or landscape) using the
aspect ratio of the
domain to be plotted, and it will plot the largest area that maintains
this ratio.
Problem
The problem buttons load default data ranges, axis orientation, and
velocity information for different problems. New problems can be added
quite easily by modifying the file tools/fidlr/xflash_defaults.pro.
It is not necessary to add a problem in order to plot a dataset.
The problem selection exists only to provide rational default values
for data ranges; these values can be overridden through the widget.
Variables
Presently the following variables can be plotted: temperature, density,
pressure, total energy, velocity magnitude, and abundance conservation
error. The latter is the difference between the sum of the nuclear
abundances in a zone and unity. The choice of problem and variable
affects the default data range chosen.
Colormap
The colormaps used by xflash differ from the standard IDL
colormaps. The first 12 colors are uniform across the colormaps and
contain standard colors (red, green, blue, white, black) that are
used to set the background color, text color, and so forth.
Plotting options
Two plotting options are currently available. The ``log'' option plots
the common logarithm of the chosen variable, while the ``show blocks''
option overlays the boundaries of the AMR block structure on the colormap
used to represent the variable. For AMR grids with many levels of
refinement, the block boundaries can completely obscure the colormap;
however, this option can be useful for verifying that resolution elements
are being placed where they are needed.
Data range
The default data range may be overridden if desired by entering a new
minimum or maximum value here. The values of the defaults are set in
tools/fidlr/xflash_defaults.pro.
Zoom
A value of -1 here indicates that the default position for the
corresponding limit is to be used. Leave each limit at -1 to plot the
entire grid, or enter different limits to plot a smaller part of the
grid.
Velocity vectors
If this box is checked, velocity arrows will be overlaid on the colormap.
The arrow lengths are scaled according to a ``typical'' velocity
magnitude, and magnitudes outside of a specified range are not plotted.
The values of the typical velocity and the clipping range are set in
tools/fidlr/xflash_defaults.pro. If the velocity vector box is
checked, the xskip and yskip options become selectable. These options
set the number of zones to skip in each direction when plotting arrows.
For highly refined meshes this can help one to avoid obscuring the
colormap with arrows.
Control buttons
Once all of the options are set, press the ``Plot'' button to produce the plot. The status of the plot appears in the status window below the buttons. If only a single plot is made (ie. only one suffix was specified), additional plots can be made without reading in the dataset again. This permits one to easily experiment with color tables, data ranges, and so forth. Once the plot is complete, press the ``Query'' button and click anywhere in the plot to probe the dataset. Mouse clicks will bring up a window showing the position of the click and the values of the hydrodynamical variables at that position.
Press the ``Quit'' button to terminate the widget.
Following is a description of each of the IDL routines included with the
FLASH IDL tools in the tools/fidlr subdirectory.
Main programs
| xflash.pro | Widget routine and event handler. |
| xflash_defaults.pro | Contains problem-specific defaults. |
| xplot_amr.pro | Main plotting routine, called by xflash. |
| read3_amr.pro | Reads block-structured AMR data from an HDF 4 file. The data are stored in common blocks. |
| read_amr_f77.pro | Reads block-structured AMR data from a Fortran unformatted file. The data are stored in common blocks. |
| def_common.pro | Defines the common blocks which hold the AMR data. Run this from the command line in order to access the AMR data structure directly (e.g., in order to pass a variable name to one of the uniform resampling routines). |
| merge_amr.pro | Resamples a given AMR data structure on a uniform grid of a given coarseness. Currently the only way to manipulate 3D data. |
| scale2_amr.pro | Resamples a given AMR data structure on a uniform grid, producing a byte array for plotting purposes. |
Plotting routines
| partvelvec.pro | Plots velocity vectors. |
| draw_blocks.pro | Plots the outline of the AMR block structure. |
| colorbar2.pro | Produces a horizontal color key. |
| vcolorbar.pro | Produces a vertical color key. |
| cmcongrid.pro | A special version of congrid used by tvimage. |
| color_gif.pro | Captures a window and saves it to a GIF file. |
| nolabel.pro | A hack to allow unlabelled axes. |
| start.pro | When run at the start of an IDL session, sets up an eight-bit X visual on a 24-bit display (when supported by the X server). |
| tvimage.pro | Similar to the IDL tv command, but allows a byte array to be drawn on the screen or directly into a Postscript file. |
| undefine.pro | Frees up memory used by AMR data structures. |
To verify that FLASH works as expected, and to debug changes in the code, we have created a suite of standard test problems. Most of these problems have analytical solutions which can be used to test the accuracy of the code. The remaining problems do not have analytical solutions, but they produce well-defined flow features which have been verified by experiments and are stringent tests of the code. The test suite configuration code is included with the FLASH source tree (in the setups/ directory), so it is easy to configure and run FLASH with any of these problems `out of the box.' Sample runtime parameter files are also included. Files containing reference results for the test suite tend to be large and so are distributed separately (via the FLASH_TEST CVS tree or in archive form as FLASH_TEST.tar). The FLASH_TEST tree also contains the runtime parameter files needed to reproduce the reference results.
Included with the test suite results is a script named flash_test which automates the process of comparing test suite results with the reference results. flash_test takes one argument, the machine type (run flash_test with no arguments for a list of available types). It automatically configures and builds FLASH for each of the test problems, builds the focu output comparison utility (Section 4.2), runs FLASH with each test problem, and uses focu to compare the results. It prepares a report for the entire suite of problems, flagging compilation problems, catastrophic runtime errors, and erroneous results. Since flash_test can be quite time-consuming to execute for the full test suite, it is generally a good idea to execute it as a batch job on the target system. Note that the tests may be run individually using the runtime parameter files provided with the FLASH test suite. Plots similar to those in this section can be produced using IDL routines provided with the test suite; these make use of the FLASH IDL tools (Section 4.3).
The Sod problem (Sod 1978) is an essentially one-dimensional flow discontinuity problem which provides a good test of a compressible code's ability to capture shocks and contact discontinuities with a small number of zones and to produce the correct density profile in a rarefaction. It also tests a code's ability to correctly satisfy the Rankine-Hugoniot shock jump conditions. When implemented at an angle to a multidimensional grid, it can also be used to detect irregularities in planar discontinuities produced by grid geometry or operator splitting effects.
We construct the initial conditions for the Sod problem by establishing a planar interface at some angle to the x and y axes. The fluid is initially at rest on either side of the interface, and the density and pressure jumps are chosen so that all three types of flow discontinuity (shock, contact, and rarefaction) develop. To the ``left'' and ``right'' of the interface we have
| (37) |
In FLASH, the Sod problem (sod) uses the runtime parameters listed in Table 1 in addition to the regular ones supplied with the code. For this problem we use the eos/gamma equation of state module and set the value of the parameter gamma supplied by this module to 1.4. The default values listed in Table 1 are appropriate to a shock with normal parallel to the x-axis which initially intersects that axis at x = 0.5 (halfway across a box with unit dimensions).
| Variable | Type | Default | Description |
| rho_left | real | 1 | Initial density to the left of the interface (rL) |
| rho_right | real | 0.125 | Initial density to the right (rR) |
| p_left | real | 1 | Initial pressure to the left (pL) |
| p_right | real | 0.1 | Initial pressure to the right (pR) |
| u_left | real | 0 | Initial velocity (perpendicular to interface) to the left (uL) |
| u_right | real | 0 | Initial velocity (perpendicular to interface) to the right (uR) |
| xangle | real | 0 | Angle made by interface normal with the x-axis (degrees) |
| yangle | real | 90 | Angle made by interface normal with the y-axis (degrees) |
| posn | real | 0.5 | Point of intersection between the interface plane and the x-axis |
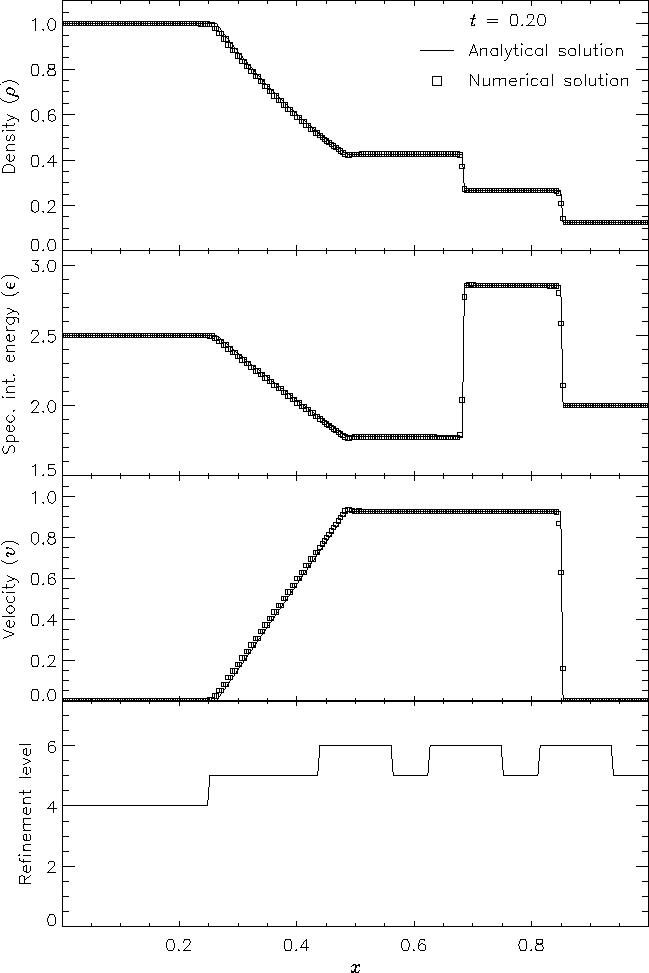
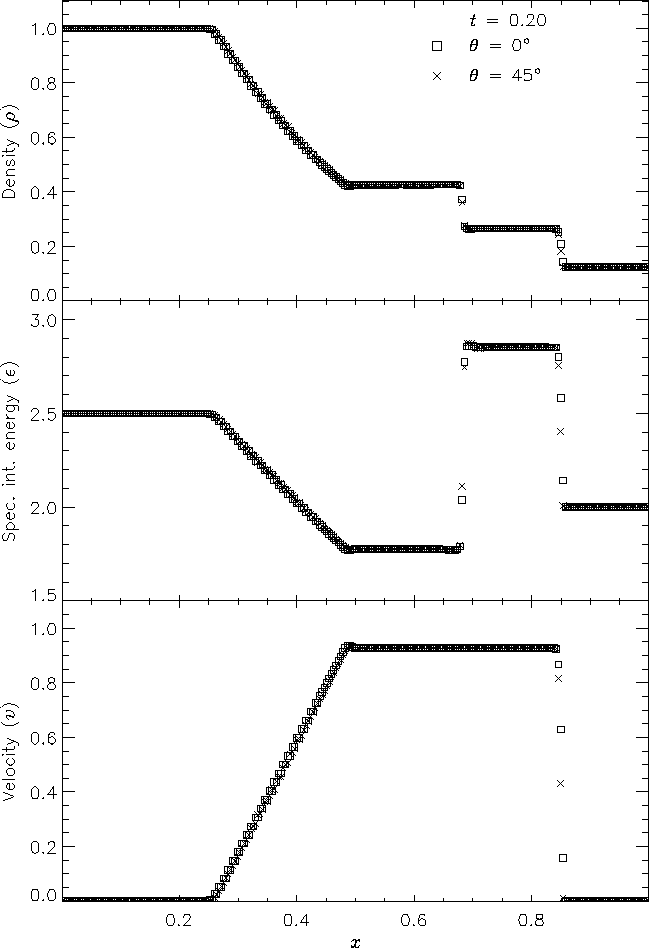
Figure 6 shows the result of running the Sod problem with FLASH on a two-dimensional grid, with the analytical solution shown for comparison. The hydrodynamical algorithm used here is the directionally split piecewise-parabolic method (PPM) included with FLASH. In this run the shock normal is chosen to be parallel to the x-axis. With six levels of refinement, the effective grid size at the finest level is 2562, so the finest zones have width 0.00390625. At t = 0.2 three different nonlinear waves are present: a rarefaction between x � 0.25 and x � 0.5, a contact discontinuity at x � 0.68, and a shock at x � 0.85. The two sharp discontinuities are each resolved with approximately three zones at the highest level of refinement, demonstrating the ability of PPM to handle sharp flow features well. Near the contact discontinuity and in the rarefaction we find small errors of about 1-2% in the density and specific internal energy, with similar errors in the velocity inside the rarefaction. Elsewhere the numerical solution is exact; no oscillation is present.
Figure 7 shows the result of running the Sod problem on the same two-dimensional grid with different shock normals: parallel to the x-axis (q = 0�) and along the box diagonal (q = 45�). For the diagonal solution we have interpolated values of density, specific internal energy, and velocity to a set of 256 points spaced exactly as in the x-axis solution. This comparison shows the effects of the second-order directional splitting used with FLASH on the resolution of shocks. At the right side of the rarefaction and at the contact discontinuity the diagonal solution undergoes slightly larger oscillations (on the order of a few percent) than the x-axis solution. Also, the value of each variable inside the discontinuity regions differs between the two solutions by up to 10% in most cases. However, the location and thickness of the discontinuities is the same between the two solutions. In general shocks at an angle to the grid are resolved with approximately the same number of zones as shocks parallel to a coordinate axis.
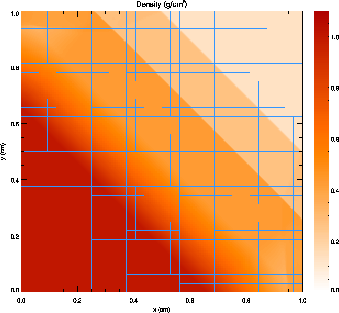
Figure 8 presents a grayscale map of the density at t = 0.2 in the diagonal solution together with the block structure of the AMR grid in this case. Note that regions surrounding the discontinuities are maximally refined, while behind the shock and discontinuity the grid has de-refined as the second derivative of the density has decreased in magnitude. Because zero-gradient outflow boundaries were used for this test, some reflections are present at the upper left and lower right corners, but at t = 0.2 these have not yet propagated to the center of the grid.
This problem was originally used by Woodward and Colella (1984) to compare the performance of several different hydrodynamical methods on problems involving strong, thin shock structures. It has no analytical solution, but since it is one-dimensional, it is easy to produce a converged solution by running the code with a very large number of zones, permitting an estimate of the self-convergence rate when narrow, interacting discontinuities are present. For FLASH it also provides a good test of the adaptive mesh refinement scheme.
The initial conditions consist of two parallel, planar flow discontinuities. Reflecting boundary conditions are used. The density in the left, middle, and right portions of the grid (rL, rM, and rR, respectively) is unity; everywhere the velocity is zero. The pressure is large to the left and right and small in the center:
| (38) |
Figure 9 shows the density and velocity profiles at several different times in the converged solution, demonstrating the complexity inherent in this problem. The initial pressure discontinuities drive shocks into the middle part of the grid; behind them, rarefactions form and propagate toward the outer boundaries, where they are reflected back onto the grid and interact with themselves. By the time the shocks collide at t = 0.028, the reflected rarefactions have caught up to them, weakening them and making their post-shock structure more complex. Because the right-hand shock is initially weaker, the rarefaction on that side reflects from the wall later, so the resulting shock structures going into the collision from the left and right are quite different. Behind each shock is a contact discontinuity left over from the initial conditions (at x � 0.50 and 0.73). The shock collision produces an extremely high and narrow density peak; in the Woodward and Colella calculation the peak density is slightly less than 30. Even with ten levels of refinement, FLASH obtains a value of only 18 for this peak. Reflected shocks travel back into the colliding material, leaving a complex series of contact discontinuities and rarefactions between them. A new contact discontinuity has formed at the point of the collision (x � 0.69). By t = 0.032 the right-hand reflected shock has met the original right-hand contact discontinuity, producing a strong rarefaction which meets the central contact discontinuity at t = 0.034. Between t = 0.034 and t = 0.038 the slope of the density behind the left-hand shock changes as the shock moves into a region of constant entropy near the left-hand contact discontinuity.
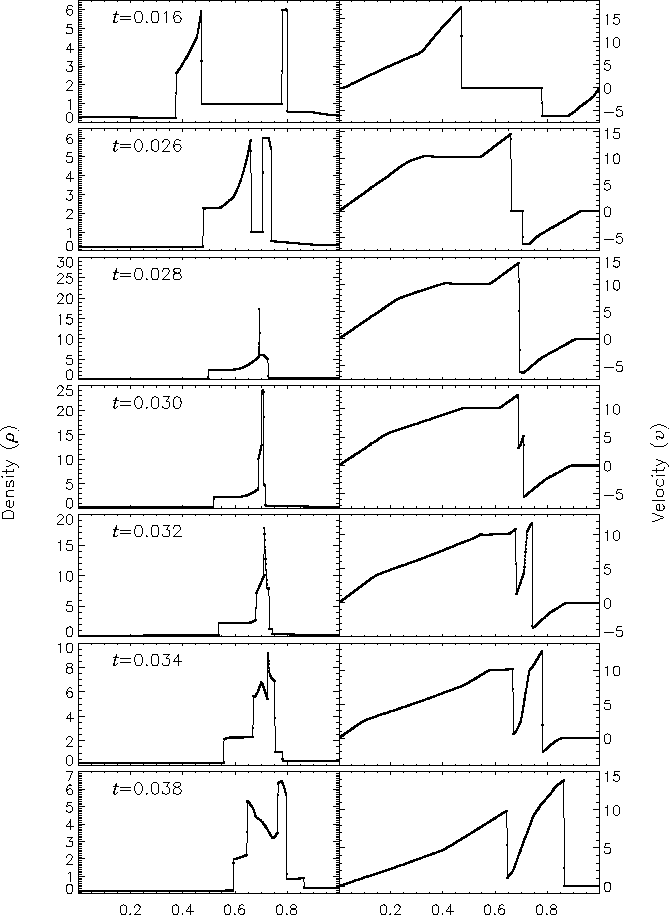
Figure 10 shows the self-convergence of density and pressure when FLASH is run on this problem. For several runs with different maximum refinement levels, we compare the density, pressure, and total specific energy at t = 0.038 to the solution obtained using FLASH with ten levels of refinement. This figure plots the L1 error norm for each variable u, defined using
| (39) |
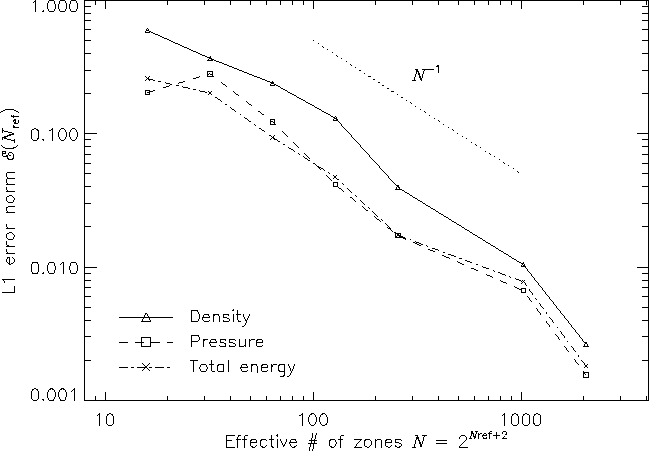
Although PPM is formally a second-order method, one sees from this plot that, for the interacting blast-wave problem, the convergence rate is only linear. Indeed, in their comparison of the performance of seven nominally second-order hydrodynamic methods on this problem, Woodward and Colella found that only PPM achieved even linear convergence; the other methods were worse. The error norm is very sensitive to the correct position and shape of the strong, narrow shocks generated in this problem.
The additional runtime parameters supplied with the 2blast problem are listed in Table 2. This problem is configured to use the perfect-gas equation of state (eos/gamma), and it is run in a two-dimensional unit box with gamma set to 1.4. Boundary conditions in the y direction (transverse to the shock normals) are taken to be periodic.
| Variable | Type | Default | Description |
| rho_left | real | 1 | Initial density to the left of the left interface (rL) |
| rho_mid | real | 1 | Initial density between the interfaces (rM) |
| rho_right | real | 1 | Initial density to the right of the right interface (rR) |
| p_left | real | 1000 | Initial pressure to the left (pL) |
| p_mid | real | 0.01 | Initial pressure in the middle (pM) |
| p_right | real | 100 | Initial pressure to the right (pR) |
| u_left | real | 0 | Initial velocity (perpendicular to interface) to the left (uL) |
| u_mid | real | 0 | Initial velocity (perpendicular to interface) in the middle (uL) |
| u_right | real | 0 | Initial velocity (perpendicular to interface) to the right (uR) |
| xangle | real | 0 | Angle made by interface normal with the x-axis (degrees) |
| yangle | real | 90 | Angle made by interface normal with the y-axis (degrees) |
| posnL | real | 0.1 | Point of intersection between the left interface plane and the x-axis |
| posnR | real | 0.9 | Point of intersection between the right interface plane and the x-axis |
The Sedov explosion problem (Sedov 1959) is another purely hydrodynamical test in which we check the code's ability to deal with strong shocks and non-planar symmetry. The problem involves the self-similar evolution of a cylindrical or spherical blast wave from a delta-function initial pressure perturbation in an otherwise homogeneous medium. To initialize the code, we deposit a quantity of energy E = 1 into a small region of radius dr at the center of the grid. The pressure inside this volume, p0�, is given by
| (40) |
| (41) |
| |||||||||||||||||||||||||
| |||||||||||||||||||
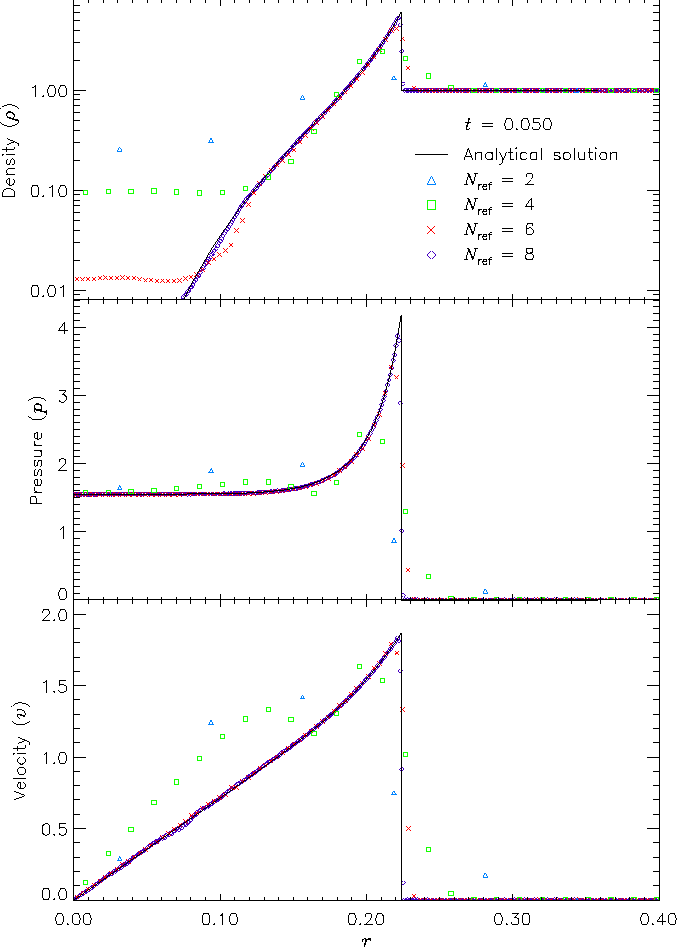
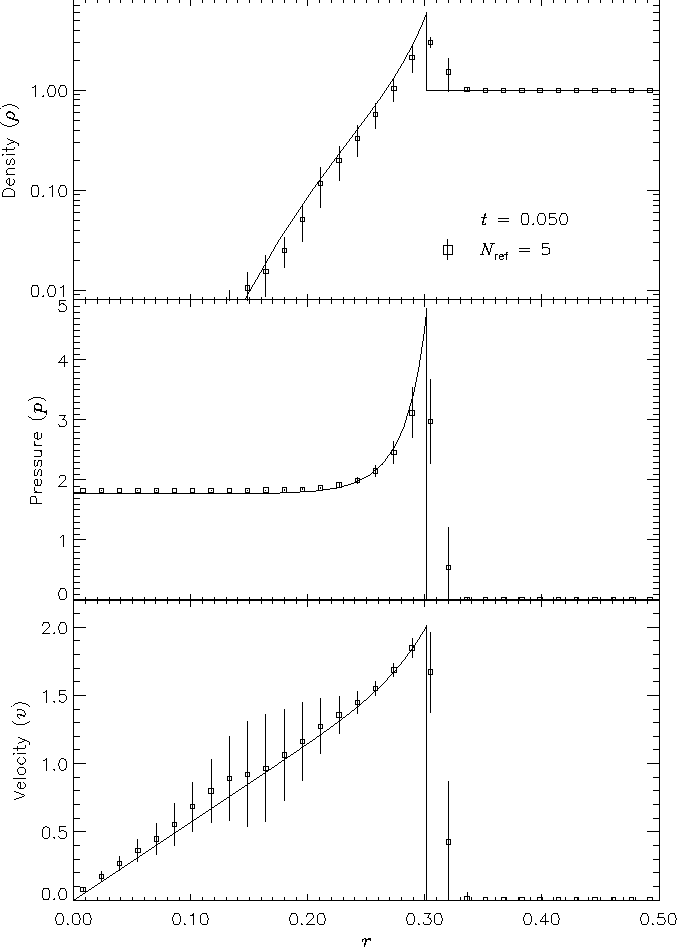
Figure 11 shows density, pressure, and velocity profiles in the two-dimensional Sedov problem at t = 0.05. Solutions obtained with FLASH on grids with 2, 4, 6, and 8 levels of refinement are shown in comparison with the analytical solution. In this figure we have computed average radial profiles in the following way. We interpolated solution values from the adaptively gridded mesh used by FLASH onto a uniform mesh having the same resolution as the finest AMR blocks in each run. Then, using radial bins with the same width as the zones in the uniform mesh, we binned the interpolated solution values, computing the average value in each bin. At low resolutions, errors show up as density and velocity overestimates behind the shock, underestimates of each variable within the shock, and a numerical precursor spanning 1-2 zones in front of the shock. However, the central pressure is accurately determined, even for two levels of refinement; because the density goes to a finite value rather than its correct limit of zero, this corresponds to a finite truncation of the temperature (which should go to infinity as r� 0). As resolution improves, the artificial finite density limit decreases; by Nref = 6 it is less than 0.2% of the peak density. Except for the Nref = 2 case, which does not show a well-defined peak in any variable, the shock itself is always captured with about two zones. The region behind the shock containing 90% of the swept-up material is represented by four zones in the Nref = 4 case, 17 zones in the Nref = 6 case, and 69 zones for Nref = 8. However, because the solution is self-similar, for any given maximum refinement level the shock will be four zones wide at a sufficiently early time. The behavior when the shock is underresolved is to underestimate the peak value of each variable, particularly the density and pressure.
Figure 12 shows the pressure field in the 8-level calculation at t = 0.05 together with the block refinement pattern. Note that a relatively small fraction of the grid is maximally refined in this problem. Although the pressure gradient at the center of the grid is small, this region is refined because of the large temperature gradient there. This illustrates the ability of PARAMESH to refine grids using several different variables at once.
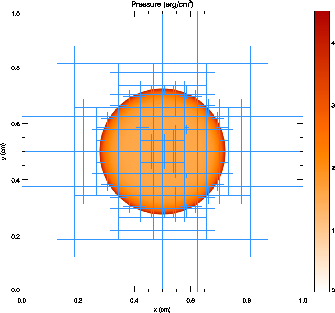
We have also run FLASH on the spherically symmetric Sedov problem in order to verify the code's performance in three dimensions. The results at t = 0.05 using five levels of grid refinement are shown in Figure 13. In this figure we have plotted the root-mean-square (RMS) numerical solution values in addition to the average values. As in the two-dimensional runs, the shock is spread over about two zones at the finest AMR resolution in this run. The width of the pressure peak in the analytical solution is about 1 1/2 zones at this time, so the maximum pressure is not captured in the numerical solution. Behind the shock the numerical solution average tracks the analytical solution quite well, although the Cartesian grid geometry produces RMS deviations of up to 40% in the density and velocity in the derefined region well behind the shock. This behavior is similar to that exhibited in the two-dimensional problem at comparable resolution.
| Variable | Type | Default | Description |
| p_ambient | real | 10-5 | Initial ambient pressure (p0) |
| rho_ambient | real | 1 | Initial ambient density (r0) |
| exp_energy | real | 1 | Explosion energy (E) |
| r_init | real | 0.05 | Radius of initial pressure perturbation (dr) |
| xctr | real | 0.5 | x-coordinate of explosion center |
| yctr | real | 0.5 | y-coordinate of explosion center |
| zctr | real | 0.5 | z-coordinate of explosion center |
The additional runtime parameters supplied with the sedov problem are listed in Table 3. This problem is configured to use the perfect-gas equation of state (eos/gamma), and it is run in a unit box with gamma set to 1.4.
In this problem we create a planar density pulse in a region of uniform pressure p0 and velocity u0, with the velocity normal to the pulse plane. The density pulse is defined via
| (44) |
| (45) |
| (46) |
The advection problem tests the ability of the code to handle planar geometry, as does the Sod problem. It is also like the Sod problem in that it tests the code's treatment of flow discontinuities which move at one of the characteristic speeds of the hydrodynamical equations. (This is difficult because noise generated at a sharp interface, such as the contact discontinuity in the Sod problem, tends to move with the interface, accumulating there as the calculation advances.) However, unlike the Sod problem it compares the code's treatment of leading and trailing contact discontinuities (for the square pulse), and it tests the treatment of narrow flow features (for both the square and Gaussian pulse shapes). Many hydrodynamical methods have a tendency to clip narrow features or to distort pulse shapes by introducing artificial dispersion and dissipation (Zalesak 1987).
| Variable | Type | Default | Description |
| rhoin | real | 1 | Characteristic density inside the advected pulse (r1) |
| rhoout | real | 10-5 | Ambient density (r0) |
| pressure | real | 1 | Ambient pressure (p0) |
| velocity | real | 10 | Ambient velocity (u0) |
| width | real | 0.1 | Characteristic width of advected pulse (w) |
| pulse_fctn | integer | 1 | Pulse shape function to use: 1=square wave, 2=Gaussian |
| xangle | real | 0 | Angle made by pulse plane with x-axis (degrees) |
| yangle | real | 90 | Angle made by pulse plane with y-axis (degrees) |
| posn | real | 0.25 | Point of intersection between pulse midplane and x-axis |
The additional runtime parameters supplied with the advect problem are listed in Table 4. This problem is configured to use the perfect-gas equation of state (eos/gamma), and it is run in a unit box with gamma set to 1.4. (The value of g does not affect the analytical solution, but it does affect the timestep.)
To demonstrate the performance of FLASH on the advection problem, we have performed tests of both the square and Gaussian pulse profiles with the pulse normal parallel to the x-axis (q = 0�) and at an angle to the x-axis (q = 45�) in two dimensions. The square pulse used r1 = 1, r0 = 10-3, p0 = 10-6, u0 = 1, and w = 0.1. With six levels of refinement in the domain [0,1]×[0,1], this value of w corresponds to having about 52 zones across the pulse width. The Gaussian pulse tests used the same values of r1, r0, p0, and u0, but with w = 0.015625. This value of w corresponds to about 8 zones across the pulse width at six levels of refinement. For each test we performed runs at two, four, and six levels of refinement to examine the quality of the numerical solution as the resolution of the advected pulse improves. The runs with q = 0� used zero-gradient (outflow) boundary conditions, while the runs performed at an angle to the x-axis used periodic boundaries.
Figure 14 shows, for each test, the advected density profile at t = 0.4 in comparison with the analytical solution. The upper two frames of this figure depict the square pulse with q = 0� and q = 45�, while the lower two frames depict the Gaussian pulse results. In each case the analytical density pulse has been advected a distance u0t = 0.4, and in the figure the axis parallel to the pulse normal has been translated by this amount, permitting comparison of the pulse displacement in the numerical solutions with that of the analytical solution.
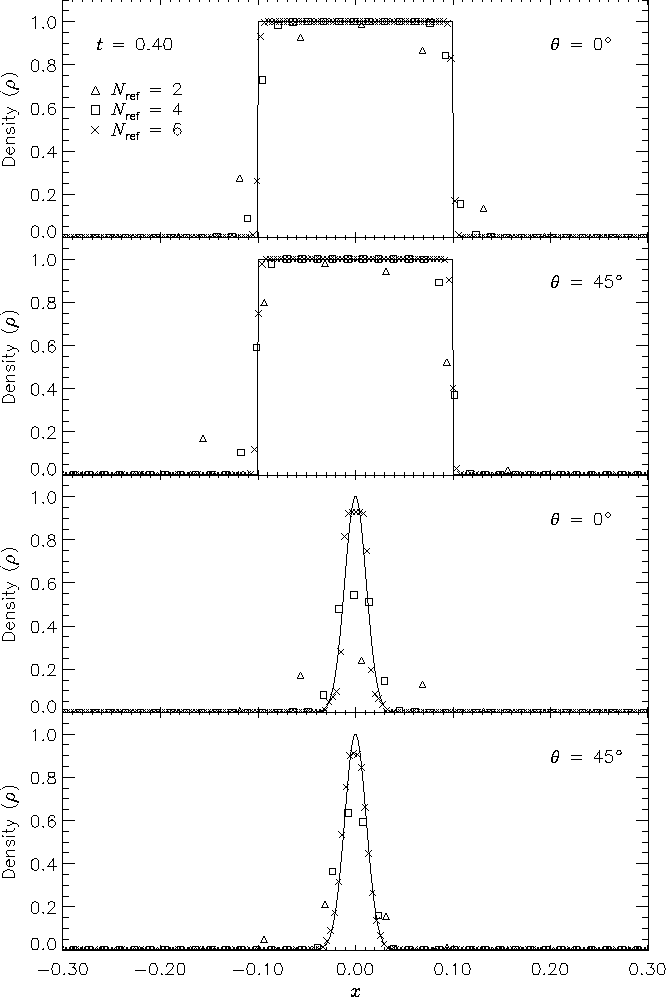
The advection results show the expected improvement with increasing AMR refinement level Nref. Inaccuracies appear as diffusive spreading, rounding of sharp corners, and clipping. In both the square pulse and Gaussian pulse tests, diffusive spreading is limited to about one zone on either side of the pulse. For Nref = 2 the rounding of the square pulse and the clipping of the Gaussian pulse are quite severe; in the latter case the pulse itself spans about two zones, which is the approximate smoothing length in PPM for a single discontinuity. For Nref = 4 the treatment of the square pulse is significantly better, but the amplitude of the Gaussian is still reduced by about 50%. In this case the square pulse discontinuities are still being resolved with 2-3 zones, but the zones are now a factor of 25 smaller than the pulse width. With six levels of refinement the same behavior is observed for the square pulse, while the amplitude of the Gaussian pulse is now 93% of its initial value. The absence of dispersive effects (ie. oscillation) despite the high order of the PPM interpolants is due to the enforcement of monotonicity in the PPM algorithm.
The diagonal runs are consistent with the runs which were parallel to the x-axis, with the possibility of a slight amount of extra spreading behind the pulse. However, note that we have determined density values for the diagonal runs by interpolation along the grid diagonal. The interpolation points are not centered on the pulses, so the density does not always take on its maximum value (particularly in the lowest-resolution case).
These results are consistent with earlier studies of linear advection with PPM (e.g., Zalesak 1987). They suggest that, in order to preserve narrow flow features in FLASH, the maximum AMR refinement level should be chosen so that zones in refined regions are at least a factor 5-10 smaller than the narrowest features of interest. In cases in which the features are generated by shocks (rather than moving with the fluid), the resolution requirement is not as severe, as errors generated in the preshock region are driven into the shock rather than accumulating as it propagates.
The problem of a wind tunnel containing a step was first described by Emery (1968), who used it to compare several hydrodynamical methods which are only of historical interest now. Woodward and Colella (1984) later used it to compare several more advanced methods, including PPM. Although it has no analytical solution, this problem is useful because it exercises a code's ability to handle unsteady shock interactions in multiple dimensions. It also provides an example of the use of FLASH to solve problems with irregular boundaries.
The problem uses a two-dimensional rectangular domain three units wide and one unit high. Between x = 0.6 and x = 3 along the x-axis is a step 0.2 units high. The step is treated as a reflecting boundary, as are the lower and upper boundaries in the y direction. For the right-hand x boundary we use an outflow (zero gradient) boundary condition, while on the left-hand side we use an inflow boundary. In the inflow boundary zones we set the density to r0, the pressure to p0, and the velocity to u0, with the latter directed parallel to the x-axis. The domain itself is also initialized with these values. For the Emery problem we use
| (47) |
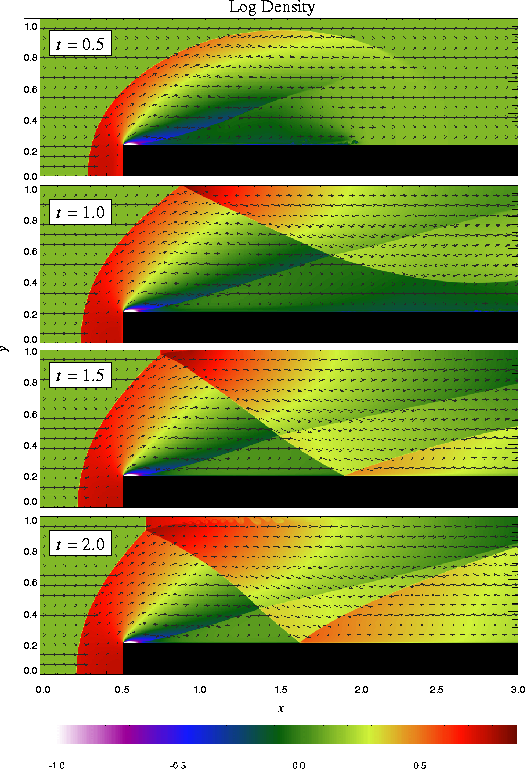
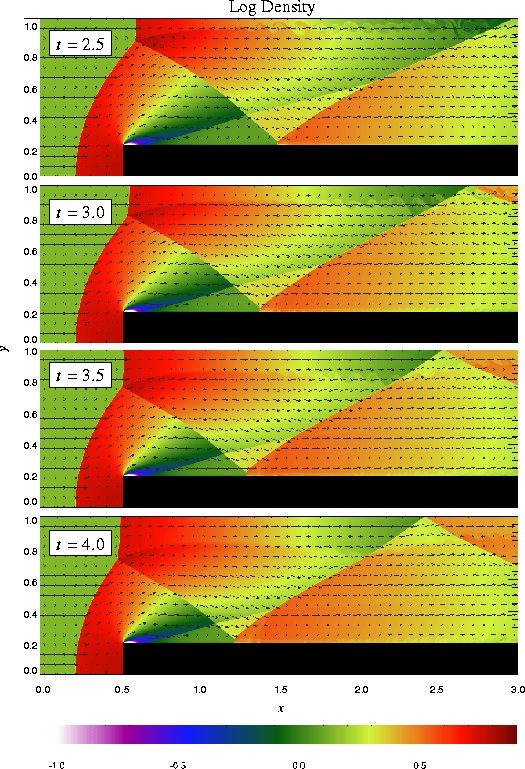
The additional runtime parameters supplied with the windtunnel problem are listed in Table 5. This problem is configured to use the perfect-gas equation of state (eos/gamma) with gamma set to 1.4. We also set xmax = 3, ymax = 1, Nblockx = 15, and Nblocky = 4 in order to create a grid with the correct dimensions. The version of divide_domain supplied with this problem adds three top-level blocks along the lower left-hand corner of the grid to cover the region in front of the step. Finally, we use xlboundary = -23 (user boundary condition) and xrboundary = -21 (outflow boundary) to instruct FLASH to use the correct boundary conditions in the x direction. Boundaries in the y direction are reflecting (-20).
Until t = 12 the flow is unsteady, exhibiting multiple shock reflections and interactions between different types of discontinuity. Figure 15 shows the evolution of density and velocity between t = 0 and t = 4 (the period considered by Woodward and Colella). Immediately a shock forms directly in front of the step and begins to move slowly away from it. Simultaneously the shock curves around the corner of the step, extending farther downstream and growing in size until it strikes the upper boundary just after t = 0.5. The corner of the step becomes a singular point, with a rarefaction fan connecting the still gas just above the step to the shocked gas in front of it. Entropy errors generated in the vicinity of this singular point produce a numerical boundary layer about one zone thick along the surface of the step. Woodward and Colella reduce this effect by resetting the zones immediately behind the corner to conserve entropy and the sum of enthalpy and specific kinetic energy through the rarefaction. However, we are less interested here in reproducing the exact solution than in verifying the code and examining the behavior of such numerical effects as resolution is increased, so we do not apply this additional boundary condition. The errors near the corner result in a slight overexpansion of the gas there and a weak oblique shock where this gas flows back toward the step. At all resolutions we also see interactions between the numerical boundary layer and the reflected shocks which appear later in the calculation.
By t = 1 the shock reflected from the upper wall has moved downward and has almost struck the top of the step. The intersection between the primary and reflected shocks begins at x � 1.45 when the reflection first forms at t � 0.65, then moves to the left, reaching x � 0.95 at t = 1. As it moves, the angle between the incident shock and the wall increases until t = 1.5, at which point it exceeds the maximum angle for regular reflection (40� for g = 1.4) and begins to form a Mach stem. Meanwhile the reflected shock has itself reflected from the top of the step, and here too the point of intersection moves leftward, reaching x � 1.65 by t = 2. The second reflection propagates back toward the top of the grid, reaching it at t = 2.5 and forming a third reflection. By this time in low-resolution runs we see a second Mach stem forming at the shock reflection from the top of the step; this results from the interaction of the shock with the numerical boundary layer, which causes the angle of incidence to increase faster than in the converged solution. Figure 16 compares the density field at t = 4 as computed by FLASH using several different maximum levels of refinement. Note that the size of the artificial Mach reflection diminishes as resolution improves.
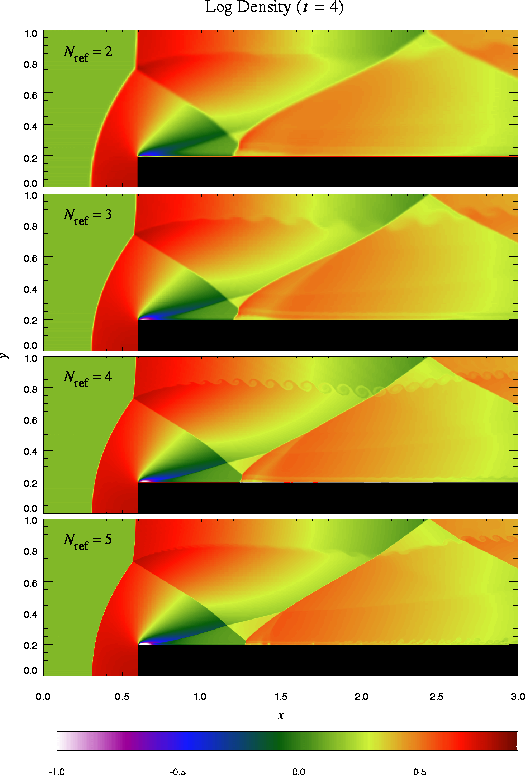
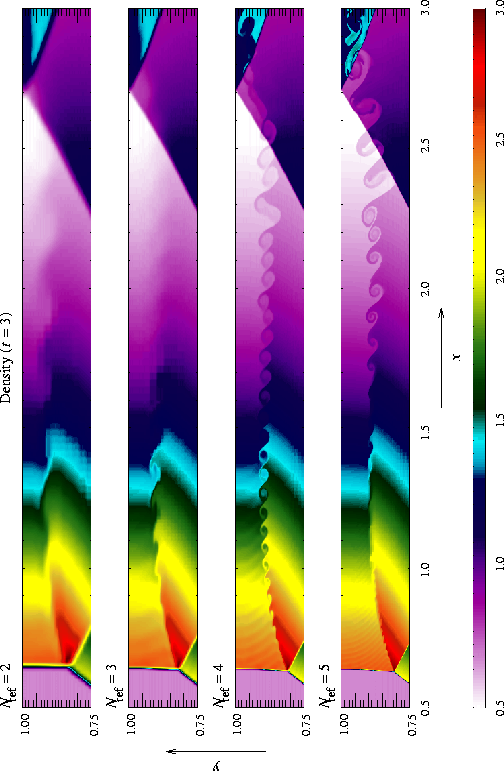
The shear zone behind the first (``real'') Mach stem produces another interesting numerical effect, visible at t = 3 and t = 4: Kelvin-Helmholtz amplification of numerical errors generated at the shock intersection. The wave thus generated propagates downstream and is refracted by the second and third reflected shocks. This effect is also seen in the calculations of Woodward and Colella, although their resolution was too low to capture the detailed eddy structure we see. Figure 17 shows the detail of this structure at t = 3 on grids with several different levels of refinement. The effect does not disappear with increasing resolution, for two reasons. First, the instability amplifies numerical errors generated at the shock intersection, no matter how small. Second, PPM captures the slowly moving, nearly vertical Mach stem with only 1-2 zones on any grid, so as it moves from one column of zones to the next, artificial kinks form near the intersection, providing the seed perturbation for the instability. This effect can be reduced by using a small amount of extra dissipation to smear out the shock, as discussed by Colella and Woodward (1984). This tendency of physical instabilities to amplify numerical noise vividly demonstrates the need to exercise caution when interpreting features in supposedly converged calculations.
Finally, we note that in high-resolution runs with FLASH we also see some Kelvin-Helmholtz rollup at the numerical boundary layer along the top of the step. This is not present in Woodward and Colella's calculation, presumably because their grid resolution is lower (corresponding to two levels of refinement for us) and because of their special treatment of the singular point.
| Variable | Type | Default | Description |
| p_ambient | real | 1 | Ambient pressure (p0) |
| rho_ambient | real | 1.4 | Ambient density (r0) |
| wind_vel | real | 3 | Inflow velocity (u0) |
FLASH is designed to be easy to configure, use, and extend. Configuration and use involve selecting particular sets of initial and boundary conditions, physics modules, and solution algorithms, as well as setting the values of parameters which control the simulation. Extension involves the addition of new problems, physics, and algorithms to suit the user's requirements. In this section we describe in some detail the structure of the FLASH source code, the format of the configuration files, and the runtime parameters and output formats available. We also discuss the steps a user needs to take in order to customize the code.
FLASH 1.0 consists of several modules which perform various high-level tasks, such as driving the code, solving the Euler equations, and solving the nuclear network equations. Figure 18 schematically represents the relationships between these modules. In this figure, a line connecting a higher block to a lower block indicates that the routines in the higher block call routines in the lower block. While most of the modules are represented by a single block, the three major functions of the driver module (initialization, evolution, and input/output) are broken out into separate blocks. This reflects the fact that the driver module included with FLASH 1.0 is oriented toward hyperbolic systems of equations. The PARAMESH library functions as the adaptive mesh refinement (AMR) module in FLASH 1.0 (see Section 2.2 for details).
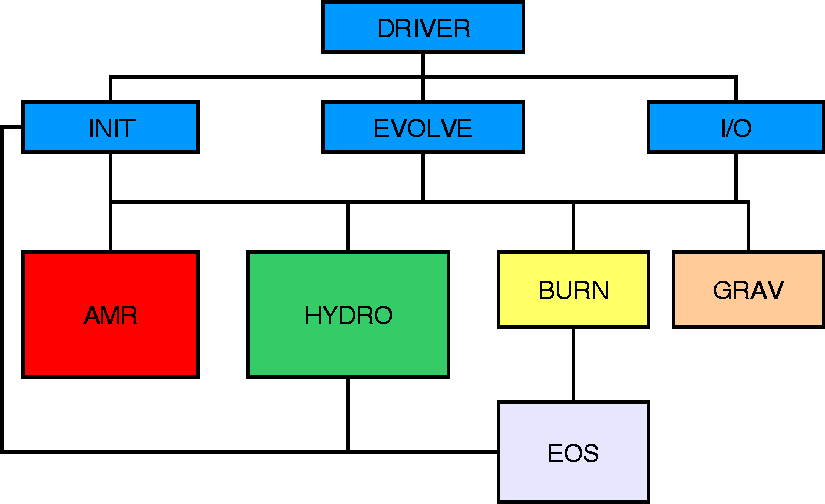
While modules in FLASH carry out generic high-level functions, often it is desirable to carry out these functions with different types of solver or choices of physics. FLASH uses sub-modules to implement this capability. A sub-module is an optional or alternative collection of routines which implements a specific instance of the general functionality represented by its parent module. Modules can have multiple sub-modules, which may or may not be mutually exclusive. Sub-modules may themselves have sub-modules. Figure 19 shows two examples of module/sub-module relationships in FLASH; one (the EOS) uses sub-modules to implement different physics, while the other (HYDRO) uses sub-modules to implement different types of solver. Code associated with a module is inherited by its sub-modules, so modules can serve as an interface layer between their sub-modules and the rest of the code. This allows different types of solver to be readily plugged in to FLASH, and when no solver for a given piece of physics is needed, the interface layer can provide empty stub routines to other parts of the code which reference the module. The following section provides more detailed information on the organization of FLASH into modules and sub-modules.
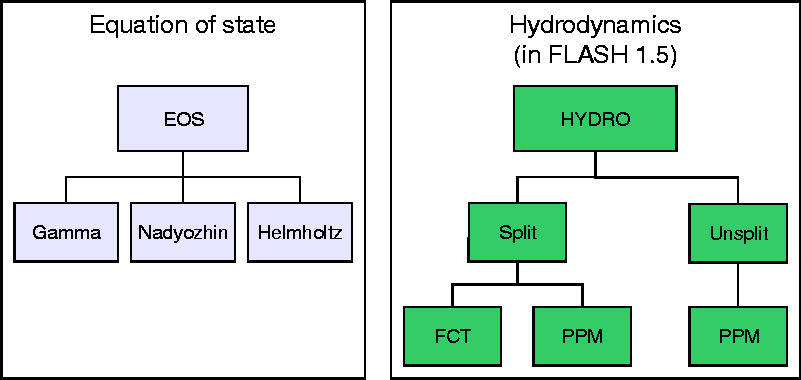
Currently data are shared among the different modules in FLASH by means of included common blocks which are part of each module. The driver module supplies the AMR mesh data structure and all runtime parameters globally to all other modules. However, variables used only within a module are declared in that module's include files, which are not included by other modules. While this mechanism does enable module data to be kept ``private'' if module programmers are disciplined, the use of global variables leaves open the possibility that new modules will attempt to redefine variables provided by the driver. In the current development version of FLASH (1.5) we are experimenting with ways to overcome this difficulty without hurting the code's performance. We expect the next version of FLASH to incorporate the use of Fortran 90 derived data types passed as arguments to modules by the driver, with no common blocks shared between modules.
The structure of the FLASH source tree is used to organize the source code into (more or less) independent code modules, as shown in Figure 20. The general plan is that source code is organized into one set of directories, while the code is built in a separate directory using links to the appropriate source files. The links are created by a source configuration script called setup, which makes the links using options selected by the user and then creates an appropriate makefile in the build directory. The user then builds the executable with a single invocation of make.
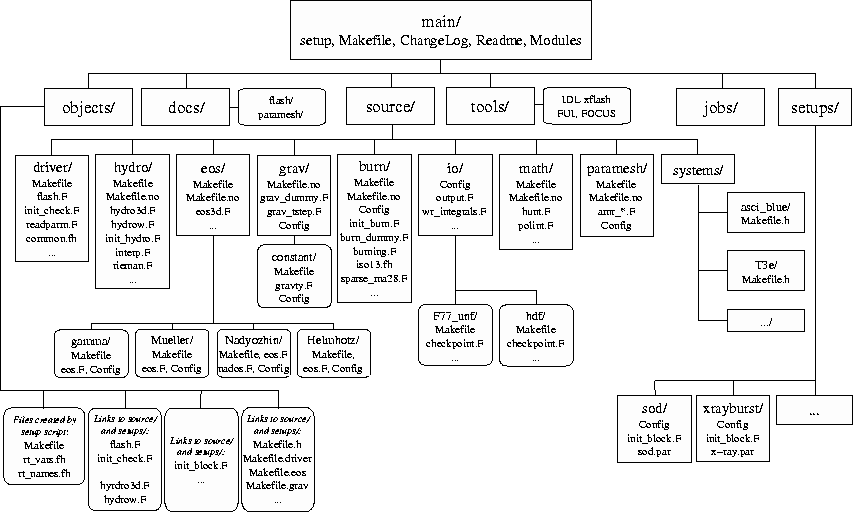
Source code for each of the different code modules is stored in subdirectories under source/. The code modules implement different physics, such as hydrodynamics, nuclear burning, and gravity, or different major program components, such as the main driver code and the input/output code. Each module directory contains source code files, makefile fragments indicating how to build the module, and a configuration file (see Section ). For each module, the makefile fragment Makefile is used when the module is included in the code, while Makefile.no is used when the module is not included (generally it builds a dummy source file containing subroutine stubs).
Each module subdirectory may also have additional sub-module directories underneath it. These contain code, makefiles, and configuration files specific to different variants of the module. For example, the hydro/ module directory can contain files which are generic to hydrodynamical solvers, while its ppm/ subdirectory contains files specific to the piecewise-parabolic method and its fct/ subdirectory contains files specific to the flux-corrected transport method. Configuration files for other modules which need hydrodynamics can specify hydro as a requirement without mentioning a specific solver; the user can then choose one solver or the other when building the code (via the Modules file; Section 6.1.3). When setup configures the source tree it treats each sub-module as inheriting all of the source code and the configuration file in its parent module's directory, so generic code does not have to be duplicated. However, makefiles in the parent directory are not inherited. Sub-modules can themselves have sub-modules, so for example one might have hydro/split/ppm and hydro/unsplit/ppm.
New solvers and new physics can be added. At the current stage of development of FLASH it is probably best to consult the authors of FLASH (see Section 7) for assistance in this. Some general guidelines for adding solvers to FLASH 1.0 may be found in Section 6.1.5 and Section 6.3.
An additional subdirectory of source/ called systems/ contains code specific to different architectures (eg., irix and t3e), each of which has its own directory under systems/. Each of these directories contains a makefile fragment called Makefile.h which is used to set machine-dependent compilation flags and so forth. In addition, special versions of any routines which are needed for each architecture are included in these directories. For example, the T3E needs a special version of getarg.f, so systems/t3e/ contains this file.
The setups/ directory has a structure similar to that of source/. In this case, however, each of the ``modules'' represents a different initial model or problem, and the problems are mutually exclusive; only one is included in the code at a time. Also, the problem directories have no equivalent of sub-modules. A makefile fragment specific to a problem need not be present, but if it is, it is called Makefile. Section 6.2 describes how to add new problem directories.
The setup script creates a directory called object/ in which the executable is built. In this directory setup creates links to all of the source code files found in the specified module and sub-module directories as well as the specified problem directory. (A source code file has the extension .c, .C, .f, .F, .fh, or .h.) Because the problem setup directory and the machine-dependent directory are scanned last, links to files in these directories override the ``defaults'' taken from the source/ tree. Hence special variants of routines needed for a particular problem can be used in place of the standard versions by simply giving the files containing them the same names.
Using information from the configuration files in the specified module and problem directories, setup creates three files needed to parse the runtime parameter file. The first two, rt_vars.fh and rt_names.fh, are Fortran include files which declare and set the default values of the parameters and associate them with character strings used for parsing the runtime parameter file. The third, rt_parms.txt, concatenates all of the PARAMETER statements found in the appropriate configuration files and so can be used as a ``master list'' of all of the runtime parameters available to the executable.
setup also creates links in object/ to all of the appropriate makefile fragments (ie. Makefile for included modules, Makefile.no for non-included modules, Makefile.h in the system-dependent directory, and Makefile in the problem directory). The link names in object/, Makefile.module, are associated with the appropriate fragments in the source directories. The setup script creates a master makefile (Makefile) in object/ which includes all of these fragments.
The master makefile created by setup creates a temporary subroutine, buildstamp.f, which echoes the date, time, and location of the build to the FLASH log file when FLASH is run. To ensure that this subroutine is regenerated each time the executable is linked, the makefile deletes buildstamp.f immediately after compiling it.
The setup script can be run with the -portable option to create a directory with real files which can be collected together with tar and moved elsewhere for building. In this case the build directory is assigned the name object_problem_machine/. Further information on the options available with setup may be found in Section 4.1.
Additional directories included with FLASH are tools/, which contains tools for working with FLASH and its output (Section 4); docs/, which contains documentation for FLASH (including this user's guide) and the PARAMESH library; and jobs/, which contains example job submission scripts for various machines.
Information about module dependencies, default sub-modules, runtime parameter definitions, and so on is stored in plain text files named Config in the different module directories. These are parsed by setup when configuring the source tree and are used to create the Fortran code needed to implement the runtime parameters, choose sub-modules when only a generic module has been specified, and to flag problems when dependencies are not resolved by some included module. In the future they may contain additional information about module interrelationships. An example Config file appears in Figure 21.
# Configuration file for hydro module
DEFAULT ppm
REQUIRES driver
REQUIRES eos
REQUIRES paramesh
PARAMETER cfl REAL 0.8 # Courant factor
The syntax of the configuration files is as follows. Arbitrarily many spaces and/or tabs may be used, but all keywords must be in uppercase. Lines not matching an admissible pattern are ignored. (Someday soon they will generate a syntax error.)
| # comment | A comment. Can appear at the end of a line. |
| DEFAULT sub-module | |
| Specify which sub-module of the current module is to be used as a default if a specific sub-module has not been indicated in the Modules file (Section 6.1.3). For example, the Config file for the eos module specifies gamma as the default sub-module. If no sub-module is explicitly included (ie. INCLUDE eos is placed in Modules), then this command instructs setup to assume that the gamma sub-module was meant (as though INCLUDE eos/gamma had been placed in Modules). | |
| REQUIRES module[/sub-module[/sub-module...]] | |
| Specify a module requirement. Module requirements can be general, not specifying sub-modules, so that module dependencies can be independent of particular algorithms. For example, the statement REQUIRES eos in a module's Config file indicates to setup that the eos module is needed by this module. No particular equation of state is specified, but some EOS is needed, and as long as one is included by Modules, the dependency will be satisfied. More specific dependencies can be indicated by specifying sub-modules. For example, eos is a module with several possible sub-modules, each corresponding to a different equation of state. To specify a requirement for, eg., the Nadozhin EOS, use REQUIRES eos/nadozhin. Giving a complete set of module requirements is helpful to the end user, because setup uses them to generate the Modules file when invoked with the -defaults option. | |
| PARAMETER name type default | |
| Specify a runtime parameter. Parameter names are unique up to 20 characters and may not contain spaces. Admissible types are REAL (or DOUBLE), INTEGER, STRING, and BOOLEAN (or LOGICAL). Default values for REAL and INTEGER parameters must be valid numbers, or compilation will fail. Default STRING values can be unquoted (in which case only the first word is recognized) or enclosed in double quotes ("). Default BOOLEAN values must be TRUE or FALSE to avoid compilation errors. Once defined, runtime parameters are available to the entire code; currently there is no mechanism for making them available only to some modules and not to others. | |
The Modules file, which is created in the FLASH root directory either by the user or by setup -defaults, is used by setup to specify the particular code modules (and sub-modules) to include. It allows the user to combine desired variants of the different code modules or to use different solution algorithms while still satisfying the dependencies of the different modules. The setup script terminates with an error if all of the dependencies of the different included modules are not satisfied by some other included module. The format of this file is similar to that of the configuration files (Section 6.1.2), but only two types of instruction are recognized:
| # comment | A comment. Can appear at the end of a line. |
| INCLUDE module[/sub-module[/sub-module...]] | |
| Specify a module (and optionally a sub-module) to include in the source configuration. If no sub-module is specified, and the configuration file for the specified module has a default sub-module, then that default will be used. For example, if the eos module is specified, then its default sub-module gamma will be used; if eos/nadozhin is specified, then the nadozhin sub-module will be used. | |
Runtime parameters are used to control the initial model, the various physics modules, and the behavior of the simulation. They are made available to FLASH via the setup configuration files described in Section 6.1.2. Their values can be set at runtime through the runtime parameter file and can be changed without requiring the user to rebuild the executable.
The runtime parameter file is a plain text file with the following format. Each line is either a comment (denoted by a hash mark, #), blank, or of the form variable = value. String values are enclosed in double quotes ("). Boolean values are indicated in the Fortran style, .true. or .false. Comments may appear at the end of a variable assignment line. An example parameter file may be found in Figure 1.
By default FLASH looks for a parameter file named flash.par in the directory from which it is run. An alternative file name can be specified as a command-line argument when FLASH is run.
In Section 6.1.5, Tables 6-11 list the runtime parameters made available by the different code modules included with FLASH. Please see Section 5 for more information on runtime parameters specific to each of the test problems supplied with FLASH.
In this section we describe in turn each of the code modules included
with FLASH, together with its available sub-modules and runtime parameters.
We also discuss the ways in which new solvers or new physics modules must
interact with each module in order to work with FLASH 1.0.
Tables 6-11
list the runtime parameters for each module.
More detailed descriptions of the algorithms included with FLASH
may be found in Section 2.
Driver module (driver)
This module contains the main program and administrative subroutines which are not tied to any specific physics module. The driver module in FLASH 1.0 is oriented toward hyperbolic problems. Hence its major functions include setting initial conditions, calling output routines (which are handled by the io module) periodically, and managing the forward time advance of the simulation (including the calculation of the timestep).
The driver module also provides the global data context for FLASH. Most physics modules access this context by including the file common.fh, which itself includes the other include files needed by the driver (hence these other files need not be explicitly included). Since common.fh and its brethren use preprocessor statements, subroutines which include it have
#include "common.fh"
as their first statement. The include files included by common.fh are:
readparm.fh, which contains declarations needed by the runtime parameter parser, including the runtime parameter buffers;
rt_vars.fh, which is generated by setup and includes all runtime parameter declarations, their default settings, and their equivalences to the runtime parameter buffers;
constants.fh, which contains declarations and settings for physical constants;
definitions.fh, which contains code constant declarations;
physicaldata.fh, tree.fh, workspace.fh, and block_boundary_data.fh, which contain constant and array declarations for the block-structured mesh data structure used by the code; and
mpif.h, which contains Fortran constant declarations for use with the Message-Passing Interface library. This file is provided as part of the MPI distribution, not with FLASH.
Users wishing to use or extend FLASH need not edit any of these files. However, the file physicaldata.fh sets two constants, maxblocks and nuc2, which are important. maxblocks sets the maximum number of AMR blocks which may be allocated to a given processor. Normally this may be set at compile time using setup -maxblocks. The second constant, nuc2, determines the number of nuclear abundances to track. The default value is 13. For problems not requiring nuclear burning, it may be desirable to set this to a smaller value in order to reduce memory usage; however, this is not required. At present nuc2 can only be changed by editing physicaldata.fh. (Note that if you wish to increase nuc2 and turn nuclear burning on, you should also increase the ionmax parameter, which is set in iso13.fh.)
| Variable | Type | Default | Description |
| basenm | string | chkpnt_ | File name prefix for checkpoint files (including the file to start from, if restart = .true.) |
| nend | integer | 100 | Maximum number of timesteps to take before halting the simulation |
| nrstrt | integer | 10000 | Maximum number of steps to take before creating a checkpoint file |
| restart | boolean | .false. | Set to .true. to restart the simulation from a checkpoint file |
| tmax | real | 1 | Maximum simulation time to advance before halting the simulation |
| trstrt | real | 1 | Maximum simulation time to advance before creating a checkpoint file |
| dtini | real | 10-10 | Initial timestep |
| small | real | 10-10 | Generic small cutoff value for positive definite quantities |
| smlrho | real | 10-10 | Cutoff value for density |
| smallp/e/t/u/x | real | 10-10 | Cutoff values for pressure, energy, temperature, velocity, and nuclear abundances |
| nsdim | integer | 2 | Grid dimensionality |
| x/y/zmin | real | 0 | Minimum x, y, and z coordinates for grid |
| x/y/zmax | real | 1 | Maximum x, y, and z coordinates for grid |
| igeomx/y/z | integer | 0 | Grid geometry in x, y, and z directions: 0=Cartesian |
| nuc | integer | 0 | Number of nuclear species to track |
| igrav | integer | 0 | If set to 1, use gravity |
| iburn | integer | 0 | If set to 1, use nuclear burning |
| CPNumber | integer | 0 | Initial checkpoint file number. If restarting from a checkpoint, set this to the number of the file to restart from |
| x/y/zlboundary | integer | -20 | Type of external boundary in the -x, -y, and -z directions: -20=reflecting, -21=outflow, -22=periodic, -23=user-defined |
| x/y/zrboundary | integer | -20 | Type of external boundary in the +x, +y, and +z directions |
| Variable | Type | Default | Description |
| iref1 | integer | 5 | Variable to use for the first AMR refinement pass. The second spatial derivative is used to select blocks for refinement. 0=none, 1=density, 2=x-velocity, 5=pressure, 6=energy, 7=temperature, 11=He abundance, 13=Ni abundance |
| iref2 | integer | 1 | Variable for the second AMR pass |
| iref3/4 | integer | 0 | Variables for the third and fourth AMR passes |
| Nblockx/y/z | integer | 1 | Number of top-level blocks in the x, y, and z directions |
The main program is contained in the file flash.F. It first reads the runtime parameter file and initializes the AMR library and physics modules. Then, depending upon the setting of the restart parameter, it calls init_from_scratch() to initialize the grid using the specified initial conditions or init_from_checkpoint() to read the initial conditions from a checkpoint file. The most important functions carried out by init_from_scratch() are the call to divide_domain(), which allocates top-level AMR blocks, and the call to init_block(), which initializes a single block (and is part of the hydro module). All problem setups must provide a version of init_block(). On the other hand, the divide_domain() subroutine provided with FLASH is fairly general and can set up a top-level mesh with Nblockx×Nblocky× Nblockz blocks. For irregular domains (such as in the windtunnel test problem), a special version of divide_domain() should be provided as part of the problem setup. (See Section 6.2 for a discussion of creating new problem setups. The files containing the replacement routines should be given the same names as the originals.)
After initialization the main program calls output_initial() (part of the io module) to write a checkpoint file containing the initial conditions and to create the scalar/global file, to which FLASH writes the total energy and other quantities at the end of each timestep.
Following the initial output in flash.F is the main evolution loop, consisting of a call to hydro_3d() (which advances the system one timestep), a call to timestep() (which determines the size of the next timestep according to constraints provided by each of the solver modules), and a call to output() (which writes to the scalar/global file and produces checkpoint files at specified intervals). In FLASH 1.0 the evolution routine hydro_3d() is part of the hydro module, which is essentially the evolution part of the PROMETHEUS code (Section 2.1). The loop terminates either when nend timesteps have been taken or when the simulation time exceeds tmax. The last things the main program does are to checkpoint the final timestep and print CPU timing information.
Currently the hydrodynamics, equation of state, gravity, and nuclear burning
routines are all called from within hydro_3d(). In FLASH 1.5 the evolution
function is taken over from hydro_3d() by a routine called evolve() which
is part of the driver module. However, since evolution in FLASH 1.0
is handled by a routine which is properly part of the hydro module,
FLASH 1.0 should always be built with hydrodynamics included.
(Alternatively, a new non-hydro physics module might supply a replacement
for hydro_3d().)
Users wishing to implement new physics modules within FLASH 1.0 should
insert calls to their solver routines in hydro_3d(). In order to access
the block-structured mesh data, global variables (such as the simulation
time and the number of processors), and runtime parameters, new module
routines should include common.fh as discussed above.
With the data structure and modularity enhancements coming in FLASH 1.5 we
expect the process of adding a new solver to become much simpler.
See Section 6.3 for further general guidelines on
adding solvers to FLASH.
Hydrodynamics module (hydro)
The hydro module solves the Euler equations of hydrodynamics (see Section 2.1). The solver included with FLASH 1.0 is directionally split and is based on the piecewise-parabolic method (PPM; Colella & Woodward 1984). Future versions will include sub-modules to handle unsplit PPM and perhaps other hydro algorithms.
As discussed in the previous section, in its current form the hydro module handles the evolution function of the driver module through the routine hydro_3d(). This routine performs one-dimensional sweeps in the x, y, and z directions, calls the nuclear burning routine (burn()), and then applies these operators in reverse order. Hydrodynamical boundary conditions are set before each 1D sweep by a call to the PARAMESH routine amr_guardcell(). For each sweep, the code cycles through the leaf-node blocks, applying the 1D hydro operator to each in turn. For a single block, hydro_3d() loops over the coordinates transverse to the sweep direction, copying rows of data from the block array to a 1D sweep array (via getrwx(), getrwy(), and getrwz()). For each row, hydrodynamical fluxes are computed with a call to hydrow(); these fluxes are then used to update zones in the interior of the block with update_soln() and in the guard cells with update_soln_boun(). Following the application of each 1D hydrodynamical operator, the hydrodynamical fluxes are corrected to ensure conservation at jumps in refinement with a call to the PARAMESH routine amr_flux_conserve(). At the end hydro_3d() tests the grid refinement with several calls to the PARAMESH amr_test_refinement() routine, then initiates changes in refinement with a call to the PARAMESH amr_refine_derefine() routine.
Three other externally visible routines in the hydro module are init_hydro(), which initializes the module by setting the hydrodynamical variables to zero; init_block(), which sets the initial conditions in a single block; and tot_bnd(), which is called by the PARAMESH guard cell filling routines when an external boundary is encountered. The latter two routines are the most important from the standpoint of users wishing to construct a new problem setup (Section ).
In addition to the global common file common.fh, the hydrodynamical
routines include three other common files: common_hydro.fh, which
contains declarations for variables specific to 1D hydrodynamical schemes
(appropriate to the operator-split implementation in FLASH 1.0);
common_ppm.fh, which contains declarations specific to the
piecewise-parabolic method; and iso13.fh, the common file exported
by the nuclear burning module and containing information on the nuclear
isotopes to track (only used by hydro_3d()).
| Variable | Type | Default | Description |
| cfl | real | 0.8 | Courant-Friedrichs-Lewy (CFL) factor; must be < 1 for stability in explicit schemes |
| PPM-specific parameters | |||
| epsiln | real | 0.33 | PPM shock detection parameter e |
| omg1 | real | 0.75 | PPM dissipation parameter w1 |
| omg2 | real | 10 | PPM dissipation parameter w2 |
| igodu | integer | 0 | If set to 1, use the Godunov method (completely flatten all interpolants) |
| vgrid | real | 0 | Scale factor for dissipative grid velocity |
| nriem | integer | 10 | Number of iterations to use in Riemann solver |
| cvisc | real | 0.1 | Artificial viscosity constant |
Equation of state module (eos)
The eos module implements the equation of state needed by the hydrodynamical and nuclear burning solvers. Three sub-modules are available in FLASH 1.0: gamma, which implements a perfect-gas equation of state; nadozhin, which implements an equation of state appropriate to partially degenerate material at nuclear densities in thermal equilibrium with radiation (Nadyozhin 1974); and helmholtz, which uses a fast Helmholtz free-energy table interpolation to handle the same physics as nadozhin (Timmes & Swesty 1999).
The primary interface routine for the eos module in FLASH 1.0 is
eos3d(), which sets the pressure in an entire block of zones by calling
the eos() routine supplied by each equation of state sub-module.
The input data for this routine are the density, temperature,
and thermal energy for the block (the block's local identification number is
passed as an argument, and the block data are referenced through the
global common blocks).
Usually eos3d() is called when these values have been changed
(e.g., by the hydro routines) and the pressure must be made consistent with
them again. The eos() routine performs this computation for a row of zones.
(Routines in the eos module include common files exported by the
hydro and burn modules. For this [and physics] reasons,
sensible use of
eos requires the hydro module [and, for nadozhin and
helmholtz, the burn module] to be included.)
Each sub-module also supplies a routine called eos_fcn() which enables
the pressure to be returned for specified values of the density, temperature,
energy, and composition, without reference to the grid.
| Variable | Type | Default | Description |
| gamma | real | 1.6667 | Ratio of specific heats for the gas (g) |
Nuclear burning module (burn)
The nuclear burning module uses a sparse-matrix semi-implicit ordinary
differential equation (ODE)
solver to calculate the nuclear burning rate and update the fluid
variables accordingly (Timmes 1999). The primary interface routines
for this module are init_burn(), which calls routines to set up the
nuclear isotope tables needed by the module; and burn(), which
calls the ODE solver and updates the hydrodynamical variables in a
single row of a
single AMR block. In addition to the global common file common.fh
and the 1D hydrodynamical common file common_hydro.fh, the burning
routines include a common file named iso13.fh which declares variables
shared within the burning module.
| Variable | Type | Default | Description |
| tnucmin | real | 1.1×108 | Minimum temperature in K for burning to be allowed |
| tnucmax | real | 1.0×1012 | Maximum temperature in K for burning to be allowed |
| dnucmin | real | 1.0×10-10 | Minimum density (g/cm3) for burning to be allowed |
| dnucmax | real | 1.0×1014 | Maximum density (g/cm3) for burning to be allowed |
| ni56max | real | 0.4 | Maximum Ni56 mass fraction for burning to be allowed |
Gravity module (grav)
The grav module sets up the gravitational field; the effects of
gravity are handled by the hydro module. In FLASH 1.0 the only
gravitational field sub-module is constant, which creates a uniform
acceleration in the x-direction. The next version of FLASH will include
a Poisson solver to compute the gravitational field due to the matter
on the computational grid. This module supplies two routines: gravty(),
which sets the gravitational acceleration in a row of zones, and
tstep_grav(), which sets the timestep constraint due to gravitation
(currently a no-op).
| Variable | Type | Default | Description |
| gconst | real | -981 | Gravitational acceleration constant (cm/s2) |
Input/output module (io)
The io module handles the output produced by FLASH, in particular the periodic checkpoints and the regular output of total energy, mass, and momentum information. It has two sub-modules, hdf, which selects the Hierarchical Data Format (HDF) for checkpoint files, and f77_unf, which selects Fortran 77 unformatted output. The HDF output produced by io/hdf is described in Section 6.1.6.
The primary interface routines for the io module are
output_initial(), which handles output just after initialization and
before the main FLASH timestep loop is entered; output(), which handles periodic
output inside the timestep loop; output_final(), which handles
output following the completion of the timestep loop; and wr_integrals(),
which computes the totals of several quantities, including energy, mass,
momentum, and so on, then writes them to an (ASC)I file named flash.dat
at the end of each timestep. The checkpoint routines provided by the two
sub-modules of io are checkpoint_wr(), which writes checkpoint files,
and checkpoint_re(), which reads them in.
PARAMESH and related modules (paramesh,
mmpi, mpi_amr)
The paramesh module supplies the adaptive mesh-refinement (AMR) library used by the rest of FLASH (MacNeice et al. 1999). It supplies a block-structured grid with factor-of-two refinement between levels, as described in Section 2.2. The mmpi and mpi_amr modules supply routines for translating the shared-memory subroutine calls used by PARAMESH into Message-Passing Interface (MPI) calls. The next version of PARAMESH, which will be included with a future version of FLASH, relies only upon MPI calls, and these modules will cease to be included. See the PARAMESH home page at
http://esdcd.gsfc.nasa.gov/ESS/macneice/paramesh/paramesh.html
for further details.
The next version of FLASH will also include a general mesh interface
layer, permitting other adaptive mesh packages to be exchanged with
PARAMESH if desired.
| Variable | Type | Default | Description |
| lrefine_max | integer | 1 | Maximum number of levels of adaptive mesh refinement (AMR) |
| lrefine_min | integer | 1 | Minimum AMR refinement level |
| nrefs | integer | 2 | Number of timesteps to advance before adjusting the grid refinement |
Math library module (math)
The math module contains miscellaneous mathematical routines which are useful in the other modules, such as a polynomial interpolation routine and a list-searching routine, both drawn from Press et al. (1992).
Currently two output formats for checkpoint files are supported by FLASH: Hierarchical Data Format (HDF) version 4 and Fortran 77 unformatted. HDF is preferred because HDF files are more portable and the format is more extensible. In this section we describe the structure of HDF checkpoint files produced by FLASH so that users can read FLASH output files into programmable visualization packages of their choosing.
With HDF, several different types of data can be combined within the same file. Data objects are specified within a file by means of character labels, and they can be read in any order using the routines provided as part of the HDF interface. See the HDF documentation for further details. Table 12 lists the data objects which FLASH writes to HDF output files. Along with the label of each object is listed its size and type (4-byte integer or 8-byte real). For reference, note that:
| tot_blocks | = | total number of blocks |
| nxb, nyb, nzb | = | zones/block in each direction |
| dimen | = | number of dimensions |
| nvar | = | number of variables |
| nfaces | = | number of faces/block ( = 2×dimen) |
| nchild | = | maximum number of child blocks per block ( = 2dimen) |
| HDF label | Variable size and type | Description |
| `total blocks' | 1 (int) | Total number of blocks in the |
| calculation | ||
| `time' | 1 (real) | Simulation time |
| `timestep' | 1 (real) | Timestep taken |
| `timers' | 7 (real) | CPU timing information: |
| 1 - guard cell filling | ||
| 2 - hydro computations | ||
| 3 - tree computations | ||
| 4 - flux computations | ||
| 5 - eos | ||
| 6 - burning | ||
| 7 - io | ||
| `number of steps' | 1 (int) | Number of steps taken |
| `refine level' | tot_blocks (int) | Refinement level of each block |
| `node type' | tot_blocks (int) | Type of node in the tree structure; |
| node type = 1 is `good data' | ||
| `gid' | nfaces + 1 + | |
| nchild×tot_blocks (int) | gid(:,iblock) contains the global | |
| block IDs of neighbors of block iblock. | ||
| The first argument gives the direction: | ||
| 1 - minimum x direction | ||
| 2 - maximum x direction | ||
| 3 - minimum y direction | ||
| 4 - maximum y direction | ||
| 5 - minimum z direction | ||
| 6 - maximum z direction | ||
| `coordinates' | dimen×tot_blocks (real) | Center coordinates of each block: |
| coord(1,iblock) : x coordinate | ||
| coord(2,iblock) : y coordinate | ||
| coord(3,iblock) : z coordinate | ||
| `block size' | dimen×tot_blocks (real) | Dimensions of each block: |
| size(1,iblock) : x dimension | ||
| size(2,iblock) : y dimension | ||
| size(3,iblock) : z dimension | ||
| `bounding box | ||
| minimum' | dimen×tot_blocks (real) | Minimum coordinate values in each |
| block: | ||
| bnd_min(1,iblock) : x minimum | ||
| bnd_min(2,iblock) : y minimum | ||
| bnd_min(3,iblock) : z minimum |
| HDF label | Variable size and type | Description | ||||||||||||||||||||||
| 'bounding box | ||||||||||||||||||||||||
| maximum' | dimen×tot_blocks (real) | Maximum coordinate values | ||||||||||||||||||||||
| in each block: | ||||||||||||||||||||||||
| bnd_max(1,iblock) : x maximum | ||||||||||||||||||||||||
| bnd_max(2,iblock) : y maximum | ||||||||||||||||||||||||
| bnd_max(3,iblock) : z maximum | ||||||||||||||||||||||||
| 'unknowns' | nvar×nxb×nyb× nzb×tot_blocks | Simulation data: unk(n,i,j,k,b) | ||||||||||||||||||||||
| contains the bth block, nth variable, | ||||||||||||||||||||||||
| zone (i,j,k). Currently n has the | ||||||||||||||||||||||||
| following interpretation: | ||||||||||||||||||||||||
|
In general, creating new sets of initial conditions is quite straightforward. Most users need only duplicate one of the existing setups and then edit it to suit their needs. Here we list the steps needed in order to create a new problem from scratch; at any step you may wish to copy a file from a similar existing setup and use it as a starting point. For example, to create a problem with plane-parallel symmetry, but in which the plane normal is adjustable, it may be helpful to start with files from the sod test problem. Similarly, for cylindrically or spherically symmetric initial conditions the sedov problem is a useful starting point.
For any files you create in the problem's setup directory with extensions .C, .c, .F, .f, .h, or .fh, the setup script will automatically create links in the object/ directory. However, they will only be compiled and linked into the executable if you create a makefile for the problem. Files such as init_block.F, which have the same name as a file in the included source directories, will simply replace the default files and do not require special treatment. For other files, create a short Makefile (call it that) in the problem setup directory with the contents
#---special files for problem---where problem is the name of your problem.
problem = list of object files
#---end special files-----
Adding new solvers (either for new or existing physics)
to FLASH is similar in some ways to adding a problem
configuration. In general one creates a subdirectory for the solver, placing
it under the source subdirectory for the parent module if the solver implements
currently supported physics, or creating a new module subdirectory if it
does not. Put the source files required by the solver into this directory,
then create the following files:
Makefile: The make include file for the module should set a macro with the name of the module equal to a list of the object files in the module. Optionally (recommended), add a list of dependencies for each of the source files in the module. For example, the burn module's make include file is
Note that if you are adding a sub-module to a module which previously had no sub-modules, you should move the Makefile from the module directory into the sub-module directory and add the new filenames to any that are already there. If the module instead already has sub-modules, copy the Makefile from one of them and do the same thing. This ensures that the source files in the module directory are built along with the sub-module. Only the Makefile in the last sub-module in a directory path is used by setup, so sub-modules should each have a separate Makefile.burn = burning.o odeint_net.o sparse_ma28.o init_burn.o tstep_burn.o
burning.o : burning.F iso13.fh
odeint_net.o : odeint_net.F
sparse_ma28.o : sparse_ma28.F
init_burn.o : init_burn.F common.fh iso13.fh
tstep_burn.o : tstep_burn.F common.fh
Makefile.no: This make include file is used by setup when it is instructed not to include the module. Sub-modules need not have a Makefile.no; their parent modules should provide one for them. The contents of Makefile.no should be
where module is the name of the module.module = module_dummy.o module_dummy.o : module_dummy.F
module_dummy.F: This file should contain stub routines to
use when the module is not included in the code. Stubs only need to
be written for routines which are visible outside the module.
If you are writing a sub-module for an existing module, make sure
the stub routine file for the parent module contains stubs for any
sub-module routines which might be visible outside the module.
Config: Create a configuration file for the module or sub-module
you are creating. All configuration files in a sub-module path are
used by setup, so in a sense a sub-module inherits its parent
module's configuration. Config should declare any runtime
parameters you wish to make available to the code when this module
is included. It should indicate which (if any) other modules
your module requires in order to function, and it should indicate
which (if any) of its sub-modules should be used as a default if
none is specified when setup is run.
The configuration file format is described in
Section 6.1.2.
Finally, if you are creating a new module (not a sub-module of an existing module), you should add the name of the module to the line beginning
in the setup script. This is a messy requirement at the moment; in the next version of FLASH we expect setup to determine for itself what modules are available.set ALLMODULES = "driver io hydro ...
This is all that is necessary to add a module or sub-module to the code. However, it is not sufficient to have the module routines called by the code! If you are creating a new solver for an existing physics module, the module itself should provide the interface layer to the rest of the code. As long as your sub-module provides the routines expected by the interface layer, the sub-module should be ready to work. However, if you are adding a new module (or if your sub-module has externally visible routines - a no-no for the future), you will need to add calls to your externally visible routines. It is difficult to give completely general guidance; here we simply note a few things to keep in mind.
If you wish to be able to turn your module on or off without recompiling the code, create a new runtime parameter (e.g., use_module) in the driver module. You can then test the value of this parameter before calling your externally visible routines from the main code. For example, the burn module routines are only called if (iburn .eq. 1). (Of course, if the burn module is not included in the code, setting iburn to 1 will result in empty subroutine calls.)
You will need to add #include "common.fh" if you wish to have access to the global AMR data structure. Since this is the only mechanism for operating on the grid data (which is presumably what you want to do) in FLASH 1.0, you will probably want to do this. An alternative, if your module uses a pre-existing data structure, is to create an interface layer which converts the PARAMESH-inspired tree data structure used by FLASH into your data structure, then calls your routines. This will probably have some performance impact, but it will enable you to quickly get things working.
You may wish to create an initialization routine for your module which is called before anything (e.g., setting initial conditions) is done. In this case you should call the routine init_module() and place a call to it (without any arguments) in the main driver routine, flash.F, which is part of the driver module. Be sure this routine has a stub available.
If your solver introduces a constraint on the timestep, you should create a routine named tstep_module() which computes this constraint. Add a call to this routine in timestep.F (part of the driver module), using your global switch parameter if you have created one. See this file for examples. Your routine should operate on a single block and take three parameters: the timestep variable (a real variable which you should set to the smaller of itself and your constraint before returning), the minimum timestep location (an integer array with five elements), and the block identifier (an integer). Returning anything for the minimum location is optional, but the other timestep routines interpret it in the following way. The first three elements are set to the coordinates within the block of the zone contributing the minimum timestep. The fourth element is set to the block identifier, and the fifth is set to the current processor identifier (MyPE). This information tags along with the timestep constraint when blocks and solvers are compared across processors, and it is printed on stdout by the master processor along with the timestep information as FLASH advances.
Since hydro_3d() handles evolution in FLASH 1.0, you will probably need to add a call to your solver in this routine. This will require you to include the hydro module when building FLASH. Alternatively, you can provide a different hydro_3d() which handles the evolution your way, but you will need to put it in a file named something other than hydro3d.F, since all source files in all top-level module directories are linked to by setup (whether they are required by the linked-to makefiles or not).
Try to limit the number of entry points to your module. This will make it easier to update it when the next version of FLASH is released. It will also help to keep you sane.
Porting FLASH to new architectures should be fairly straightforward for most Unix or Unix-like systems. For systems which look nothing like Unix, or which have no ability to interpret the setup script or makefiles, extensive reworking of the meta-code which configures and builds FLASH would be necessary. We do not treat such systems here; rather than do so, it would be simpler for us to do the port ourselves. The good news in such cases is that, assuming that you can get the source tree configured on your system and that you have a Fortran 90 compiler and the other requirements discussed in Section 3, you should be able to compile the code without making too many changes to the source. We have generally tried to stick to standard Fortran 90, avoiding machine-specific features.
For Unix-like systems, you should make sure that your system has csh, a make which permits included makefiles, awk, and sed. You will need to create a directory in source/systems/ with the name of your machine/OS type. At a minimum this directory should contain a makefile fragment named Makefile.h. The best way to start is to copy one of the existing makefile fragments (eg. for irix) to your new directory and modify that. Makefile.h sets macros which define the names of your compilers, the compiler and linker flags, the names of additional object files needed for your machine but not included in the standard source distribution, and additional shell commands (such as file renaming and deletion commands) needed for processing the master makefile.
For most Unix systems this will be all you need to do. However, in addition to Makefile.h you may need to create machine-specific subroutines which override the defaults included with the main source code. As long as the files containing these routines duplicate the existing routines' filenames, they do not need to be added to the machine-dependent object list in Makefile.h; setup will automatically find the special routine in the system-specific directory and link to it rather than to the general routine in the main source directories.
An example of such a routine is getarg(), which returns command-line arguments and is used by FLASH to read the name of the runtime parameter file from the command line. This routine is not part of the Fortran 90 standard, but it is available on many Unix systems without the need to link to a special library. However, it is not available on the Cray T3E; instead, a routine named pxfgetarg() provides the same functionality. Therefore we have encapsulated the getarg() functionality in a routine named get_arguments(), which is part of the driver module in a file named getarg.f. The default version simply calls getarg(). For the T3E a replacement getarg.f calling pxfgetarg() is supplied. Since this file overrides a default file with the same name, getarg.o does not need to be added to the machine-dependent object list in source/systems/t3e/Makefile.h.
FLASH is still under active development, so many desirable features have not yet been included (eg., self-gravity, thermal conduction, radiation transport, etc.). Also, you will most likely encounter bugs in the distributed code. A mailing list has been established for reporting problems with the distributed FLASH code. To report a bug, send email to
flash_bugs@mcs.anl.gov
giving your name and contact information and a description of the procedure you were following when you encountered the error. Information about the machine, operating system (uname -a), and compiler you are using is also extremely helpful. Please do not send checkpoint files, as these can be very large. If the problem can be reproduced with one of the standard test problems, send a copy of your runtime parameter file and your setup options. This situation is very desirable, as it limits the amount of back-and-forth communication needed for us to reproduce the problem. We cannot be responsible for problems which arise with a physics module or initial model you have developed yourself, but we will generally try to help with these if you can narrow down the problem to an easily reproducible effect which occurs in a short program run and if you are willing to supply your added source code.
At present no address has been set up for general development questions and comments, but we expect this to change in the near future. Documentation (including this user's guide) and support information for FLASH can be obtained on the World Wide Web at
http://www.flash.uchicago.edu/flashcode/